Abstract
Regulation between protein kinases is critical for the establishment of signaling pathways/networks to 'orchestrate' cellular processes. Besides posttranslational phosphorylation, alternative pre-mRNA splicing is another way to control kinase properties, but splicing regulation between two kinases and the effect of resulting variants on cells has barely been explored. Here we examined the effect of the protein kinase A (PKA) pathway on the alternative splicing and variant properties of the Ca++/calmodulin-dependent protein kinase kinase 2 (CaMKK2) gene in B35 neuroblastoma cells. Inclusion of the exon 16 of CaMKK2 was significantly reduced by H89, a PKA selective inhibitor. Consistently, overexpressed PKA strongly promoted the exon inclusion in a CaMKK2 sequence-dependent way in splicing reporter assays. In vitro, purified CaMKKβ1 variant proteins were found to be kinase-active. In cells, they were differentially phosphorylated by PKA. In RNA interference assays, CaMKKβ1 was found to be essential for forskolin-induced neurite growth. Interestingly, overexpression of the variant without exon 16 (-E16) promoted neurite elongation while the other one (+E16) promoted neurite branching; in contrast, reduction of the latter one enhanced neurite elongation. Moreover, the variants are differentially expressed and the exon 16-containing transcripts highly enriched in the brain, particularly the cerebellum and hippocampus. Thus, PKA regulates the alternative splicing of CaMKK2 to produce variants that differentially modulate neuronal differentiation. Taken together with the many distinct variants of kinases, alternative splicing regulation likely adds another layer of modulation between protein kinases in cellular signaling networks.
Introduction
There are more than 500 protein kinase-encoding genes in the human genome.Citation1–Citation3 Regulation between the kinases forms signaling pathways and networks to coordinate important cellular processes such as gene expression and cell differentiation. While the most well-studied way of regulation has been direct phosphorylation,Citation3 accumulating evidence indicates that the alternative splicing of pre-mRNA transcripts also alter kinase functions. For example, differential catalytic activities were shown for the CDK2 and the AMP-dependent protein kinase variants.Citation4–Citation8 Selective interaction with transcription factors was observed for the JNK and protein kinase C variants.Citation9–Citation12 Moreover, various differences have been shown for the splice variant products of the CaMKII genes, including effects on cardiac muscle contraction and frequency-dependent response to Ca2+ oscillation,Citation13,Citation14 targeting to the sarcoplasmic reticulum,Citation15 and interaction with actin.Citation16 In another case, the short form of the LKB1 variant kinase is specifically essential for spermatid maturation.Citation17,Citation18 These observations indicate that, as for most other eukaryotic genes, alternative splicing is also used by protein kinase-coding genes to diversify enzyme activities, interaction with other critical proteins and consequently to impact cellular functions at higher levels. Furthermore, there are also numerous reports supporting the control of alternative splicing by protein kinases,Citation19–Citation26 but whether kinases of different pathways cross-regulate through alternative splicing remains largely unexplored.
The adenosine 3′,5′-cyclic monophosphate (cAMP) and the Ca2+ signaling pathways crosstalk through direct PKA phosphorylation of the Ca2+/calmodulin-dependent protein kinase kinases α (encoded by the CaMKK1 gene) leading to inhibition of the CaMK pathway by PKA.Citation27–Citation31 In a previous report, we found that PKA repressed splicing through an exonic element of CaMKK2 exon 16 in a heterologous exon.Citation32 We also noticed that endogeneous CaMKK2 splicing was regulated by cpt-cAMP and forskolin in rat pheochromocytoma (PC12) cells.Citation32 These evidence implied that the alternative splicing of the CaMKK2 gene was regulated by PKA; however, it was unclear whether the endogeneous PKA was essential for that regulation and whether the regulated variants could have any differential effect on cells.
In this report, we identify the endogeneous PKA pathway as an essential regulator of CaMKK2 splicing in rat B35 neuroblastoma cells as well as the differential effects of the regulated splice variants on forskolin-induced neurite growth.
Results
Cell-dependent regulation of CaMKK2 exon 16 by forskolin and the essential role of the PKA pathway in rat B35 neuroblastoma cells.
In our previous experiments using PC12 cells,Citation32 the level of the exon 16-containing transcripts of CaMKK2 () was inhibited by cpt-cAMP and forskolin, which enhanced the phosphorylation of a PKA target serine in these cells,Citation33 implying the regulation of CaMKK2 splicing by PKA. However, surprisingly, later we found that the forskolin effect was not blocked by a PKA inhibitor H89 in follow-up experiments (p > 0.05, ). A src kinase inhibitor PP2 slightly inhibited the forskolin effect (p < 0.05) but with no significant difference from H89. Thus, these observations suggest that mainly other pathways downstream of forskolin are involved in the splicing regulation of CaMKK2 in the PC12 cells. We then examined more cell lines and found that H89 had significant inhibitory effect on the relative level of exon 16-containing transcripts in B35 neuroblastoma cells (, lanes 3–8). In these cells, cAMP or forskolin did not inhibit but moderately enhanced the exon 16 level, where the small effect could be due to high basal PKA activities (, lanes 7–8). Importantly, H89 clearly reduced the relative level of exon 16-containing transcripts (lanes 4 and 6), suggesting that the PKA pathway plays an essential role in the B35 cells. Thus, forskolin appears to have cell-dependent effect on the splicing of the CaMKK2 transcripts through different kinase pathways. Here we will focus on the effect in B35 cells, where the endogeneous PKA pathway is required for the regulation of CaMKK2 splicing.
To verify that PKA regulates exon 16 usage, we used mini-gene splicing reporter assays, as we have done previously in references Citation32 and Citation34–Citation36. With this assay, we have identified from exon 16 a heptamer element that, when transferred to a heterologous exon, mediates splicing repression by PKA. For PKA promotion of the exon 16 inclusion, we reasoned that the repressor element effect could be overcome by other elements/sequences of CaMKK2, which could be present in a reporter covering more exon 16 and flanking intron sequences. We thus cloned a 720 bp DNA fragment containing exon 16 with its partial flanking introns in a splicing reporter minigene to examine the exon inclusion ().
To obtain a clear effect by PKA without high basal activities of the pathway in B35 cells, we carried out the reporter assays in HEK293T cells (). Semi-quantitative RT-PCR of the spliced products of the reporter, coexpressed with a kinase-dead Flag-PKA mutant (Mut) or kinase-active Flag-PKA (WT),Citation32,Citation37 indicates that the active PKA significantly enhanced the inclusion of the exon 16 (lanes 2–4) from about 4% to 20%, a 5-fold increase. In contrast, the kinase active PKA did not change the inclusion of two other exons similarly cloned into the same vector (lanes 5–8). Therefore, the 720 bp CaMKK2 sequence is specifically required to mediate PKA-enhanced inclusion of the exon 16.
PKA enhancing effect of exon 16 in the mini-gene was also observed in B35 cells but to a much less extend (data not shown), likely due to the high basal PKA activity in these cells.
Taken together, the inhibitor assay of the endogenous CaMKK2 and the mini-gene splicing reporter assay () support that the exon 16 inclusion is upregulated by PKA in these B35 cells.
The regulated CaMKK2 variant proteins are kinase-active and are differentially phosphorylated by PKA.
To explore functional consequences of the regulated splicing of CaMKK2, we first examined whether the resulting variant kinases are indeed active and whether they have different biochemical properties. In a previous report in reference Citation38, autophosphorylation of the CaMKK2 variants and their phosphorylation of CaMKIV were examined but the amount of proteins used in those kinase reactions were not shown for each sample, making it difficult to conclude whether the variants have the same or different kinase activities. To obtain conclusive evidence for the variant activities, we constructed plasmids to express Myc-tagged CaMKK2 proteins, Myc-CaMKK2 −E16 and Myc-CaMKK2 +E16, in HEK293T cells (). We then carried out in vitro kinase assays using immunoprecipitated proteins with their input amount monitored by western blots. The result indicated that the two variants autophosphorylated and phosphorylated CaMKIV in a Ca2+/CaM-dependent way ( and C). After the radioactive Citation32P-signals were normalized to input protein levels, the two variants showed similar activities in both overall autophosphorylation level and their phosphorylation level of CaMKIV in these assays.
PKA phosphorylation of CaMKKα has been shown to play an important role in the crosstalk between the PKA and CaMKK pathways.Citation27–Citation31 We thus examined whether the CaMKK2 variant proteins were also phosphorylated directly by PKA in in vitro phosphorylation assays. The result showed that both variants were similarly phosphorylated by PKA directly ().
To see whether the two CaMKK2 variants are phosphorylated differentially by PKA in cells, we carried out phosphopeptide mapping of the variant proteins coexpressed with the Flag-PKA in HEK293T cells. The two Myc-CaMKK2 proteins each displayed seven prominent 32P-labelled spots with several minor ones (). In contrast, the Flag-PKA mutant-coexpressed Myc-CaMKK2 +E16 sample did not show similar spots (data not shown). Interestingly, the Myc-CaMKK2 +E16 protein showed one additional spot (#8, circled). Moreover, spot #3 appeared consistently weaker in +E16 samples (40% ±4 of the −E16 samples, n = 4). Therefore, the two Myc-CaMKK2 variant proteins are differentially phosphorylated by PKA.
Moreover, in dual luciferase reporter assays using a cAMP/calcium-responsive element (CRE)-driven reporter, both variants have similar basal activities on transcription in B35 cells ().
Taken together, the biochemical properties of the CaMKK2 variant proteins demonstrate that they are enzymatically and transcriptionally active and are differentially phosphorylated by PKA.
CaMKK2 protein is localized to neurites and required for forskolin-induced neurite growth in B35 cells.
Since these variants are kinase active, exhibit differential phosphorylation by PKA and moreover, their relative levels through splicing are regulated by PKA, we tried to examine whether they have different effects on forskolin-induced neurite growth.Citation33 For this purpose, we first examined the presence and role of CaMKK2 protein in neurites upon forskolin treatment.
Forskolin induced neurite outgrowth with some branches (, lower left) in B35 cells. In unstimulated cells, CaMKK2 is predominantly nuclear by immunostaining (upper part, middle), as in the brain,Citation39 but unlike its homologue CaMKKα, which is mainly cytoplasmic.Citation40,Citation41 Upon forskolin stimulation, more CaMKK2 was found in the cytoplasm and neurites (lower part, middle). Under a higher magnification, CaMKK2 could be seen in bright foci in the neurite stem (). This observation suggests that the CaMKK2 protein is present in neurites during forskolin-induced neurite growth of B35 cells.
To see whether CaMKK2 is required for the neurite growth, we transfected an empty vector pGIPz or a plasmid expressing short hairpin RNAs (shKK2) against the CaMKK2 mRNA into B35 cells and examined the effect on neurite length and branching upon forskolin treatment (). These plasmids also express a turboGFP protein to track shRNA expression. By comparing the GFP- and non-GFP-expressing cells (), we were able to determine that the shKK2 expression significantly reduced the immunoreactivity of CaMKK2 to about 27%, while the vector pGIPz did not have a significant effect.
Forskolin induced several primary neurites per cell. The longest one was measured for each cell as its neurite length. In the vector-transfected cells, forskolin induced neurite growth to about 72 µm (±2.1, mean ± SEM, same as for the following neurite lengths, n = 181) within 7 hours ( and E), compared to the 34 µm in cells treated with vehicle control ethanol (ETOH). In contrast, in cells transfected with the shKK2-expressing plasmid, the neurite lengths were only 46 µm (±1.8, n = 162) and 27 µm in the forskolin- and ethanol-treated cells, respectively. Therefore, knocking down CaMKK2 protein significantly reduces the forskolin-induced neurite growth (p < 0.001). Similarly, the primary order neurites induced by forskolin were significantly reduced by shKK2 expression from 2.7 (±0.08) to 1.9 (±0.06) per cell (p < 0.001). CaMKK2 is thus likely essential for the forskolin-induced neurite growth and branching in B35 cells.
The regulated CaMKK2 variants exert differential effects on forskolin-induced neurite growth of B35 cells.
To investigate whether these two variants have differential effects on the neurite growth, we expressed the Myc-CaMKK2 variant proteins in B35 cells and examined their effects on neurite length and branches upon forskolin-induction ().
Both variants were found in neurites (). In cells expressing an EGFP protein, forskolin induced neurite growth to 77.7 µm (±1.4, n = 370) after 7 h, compared to 48.1 µm (±1.8, n = 106) in ethanol-treated cells or similar lengths of these green cells without forskolin stimulation (). The forskolin effect was increased significantly by expression of Myc-CaMKK2 −E16, to 95.2 µm (±1.6, n = 311, p = 1.1E-15). In contrast, the expression of Myc-CaMKK2 +E16 barely exerted any effect on the neurite length (81.7 ± 1.5 µm, n = 345), compared to the expression of EGFP (p = 0.05) (). The most significant difference between these variants lies between the 75 to 100 µm range (), with about 20% difference in cell populations at a given point of neurite length. Thus Myc-CaMKK2 −E16, but not Myc-CaMKK2 +E16, significantly enhances forskolin-induced neurite elongation of B35 cells.
To see whether the variant effect on neurite elongation and their neurite localization are also differentially controlled by the PKA pathway, we treated the cells with the PKA inhibitor H89 prior to the addition of forskolin. The enhancing effect on neurite elongation by the −E16 variant was completely abolished by H89 while the neurite length of the +E16 cells were not affected (, lower part, and C). In the neurites grown out, both variant proteins can still be clearly identified without visible difference in immunostaining. Therefore, the PKA phosphorylation is likely required for the enhancing effect of the Myc-CaMKK2 −E16 variant on forskolin-induced neurite growth but not the subcellular and neurite localization of the variants in B35 cells.
In addition to neurite elongation, we also noticed differences in neurite branching between forskolin-induced cells expressing the two variants respectively (). For the EGFP-expressing cells, the number of primary, secondary and higher order branches per cell were 3.0 (±0.06, mean ± SEM, same as follows for branches), 0.9 (±0.05), 0.2 (±0.03), respectively (n = 297). For Myc-CaMKK2 −E16-expressing cells, these numbers were: 2.8 (±0.06), 0.8 (±0.05) and 0.13 (±0.03), respectively, with significantly less higher order branches (n = 284). However, for the Myc-CaMKK2 +E16-expressing cells, the numbers were 3.2 (±0.06), 1.1 (±0.06) and 0.4 (±0.06), respectively (n = 288), significantly higher than either the EGFP or −E16 sample, particularly in higher order branches. Therefore, compared to Myc-CaMKK2 −E16, Myc-CaMKK2 +E16 enhances neurite branching, an effect that was also observed by others for both cAMP and forskolin.Citation42,Citation43
To see whether specific disturbance of the level of the endogeneous variants regulate neurite growth, we first tested 2′-OME RNA oligos complementary to the upstream 3′ or downstream 5′ splice sites, as reported in splicing assays in cells,Citation44 for their inhibitory effect on exon 16 inclusion upon transfection into B35 cells. We found that the 5′ splice site oligo had specific effect at 200 nM () and thus used this one to examine the neurite growth. In this experiment, B35 cells were non-transfected or transfected with either the 5′ splice site CaMKK2 oligo or a control oligo derived from the human ATM gene with no perfect match with rat sequences. The relative ratio of the +E16 to −E16 transcripts were specifically reduced about 50% by the CaMKK2 oligo (from 0.64 to 0.37) but only slightly affected by the ATM oligo ().
Cells transfected with or without these oligos were stained with phalloidin-TRITC for neurite measurement. Since all cells were stained, only those that were clearly isolated from others were measured for their neurites. Their neurite lengths are thus generally shorter () than the cells measured above (). However, under the same measuring conditions, they should allow us to compare the differences between the effects of the two RNA oligos.
According to this measurement, the neurite length of the nontransfected cells were 27 µm and 45 µm with ethanol and forskolin treatment, respectively ( and H). In the CaMKK2 oligo-transfected cells, these lengths were 47.5 µm (±1.9, n = 110) and 76.3 µm (±2.5, n = 91), respectively. In contrast, in the control oligo-transfected cells, these lengths were only 27 µm (±0.9, n = 112) and 46.0 µm (±1.3, n = 112), similar to the non-transfected ones. Moreover, the number of secondary neurites in the CaMKK2 oligo-treated cells was also significantly less than in the control-oligo treated cells (). Therefore, the forskolin-induced neurite length and branches in B35 cells are highly sensitive to disturbance of the endogeneous CaMKK2 variant ratios, consistent with the observed overexpression effect.
The exon-16 containing variant is highly enriched in the brain.
Previous reports have shown that the CaMKK2 gene is mainly expressed in the brain and the exon 16-containing transcripts are differentially spliced among brain tumor-derived cell lines.Citation38,Citation45 But it is not clear which splice variants are enriched in the brain. We then examined the variants in RNA samples of different regions of adult rat brain and other tissues using RT-PCR. The CaMKK2 transcripts were only weakly detectable in the lung but strongly expressed in the brain (, lanes 2 and 6–9), with barely detectable levels in the other tissues (lanes 3–5). Interestingly, in the brain samples, exon-16 containing transcripts are the predominant products with high levels in the cerebellum (99%, lane 6) and hippocampus (89%, lane 7), and there are no exon-14 skipped transcripts. The latter was only detectable in the lung. Thus the alternative exons of CaMKK2 are differentially included in the transcripts among different tissues and the exon-16 containing transcripts are highly enriched in the brain.
In summary, these data demonstrate that PKA controls the exon 16 of the CaMKK2 gene to produce variant proteins with differential effect on neurite elongation and branching. Together with their differential expression among tissues, particularly among different regions of the brain, the PKA regulation of exon 16 usage provides differential combinations of the CaMKK2 variants in a tissue and cell signal-dependent way, likely to modulate two important aspects of neurite growth: elongation and branching.
Discussion
Regulation between protein kinases are usually through phosphorylation. In this report, we provide an example where the alternative splicing of one kinase gene can also be cross-regulated by another kinase of a different pathway to produce variants with differential effects on cell differentiation.
Several labs have observed that the cAMP/PKA pathway inhibits the CaMK kinase pathway through direct phosphorylation of CaMK kinases.Citation27–Citation31 The data here show that PKA also regulates CaMKK2 splicing and the resulting splice variants exert differential effects on neurite growth. The regulation appears to increase the proportion of the CaMKK2 +E16 variant that promotes neurite branching. The regulation thus likely participate in balancing the elongation and branching effects by forskolin on neuronal differentiation. Based on these data, the regulation of CaMKK2 by PKA through alternative splicing during neurite growth likely adds another way of cross-talk between these pathways ().
The molecular basis of the differential effects of the two variants on neurite growth is not known. The variants exhibit differential phosphorylation and the phosphorylation is likely required for the specific enhancing effect on neurite elongation by the variant without E16 (−E16) ( and C). For the +E16 variant, we have mutated the potential PKA targets threonine 522 and serine 559 in the COOH terminal but have not observed clear changes in phosphorylation in peptide mapping assays (data not shown). The extra phosphopeptide #8, whose phosphorylation is dependent on the exon 16 peptide, could thus be within the common region of the variants. Identification of the phosphorylation sites of these variants will help elucidate the role of phosphorylation in the control of their properties.
Their differential phosphorylation by PKA does not seem to exert a different effect on transcription of a CRE-luciferase reporter () or their localization in neurites (). We have also tested the reporter under depolarization conditions but did not detect any difference in their activities either (data not shown). While the luciferase assays could be complicated by the high basal activities of PKA pathway ( and lanes 7–8) and promoter choice, it is also possible that other properties, such as interaction of CaMKK2 with other proteins in the neurites are effected by the differential phosphorylation.
The control of the CaMKK2 exon 16 by forskolin and cpt-cAMP appears highly complex as for most mammalian exons.Citation46,Citation47 In PC12 cells, the stimulators repress exon 16 inclusion,Citation32 but in B35 cells, they enhance it. This cell-dependent opposite effect by forskolin on the same exon is likely through different signaling pathways (): PKA in B35 cells and a mainly undetermined pathway in PC12 cells.
A “CAAAAAA” heptamer motif of the exon 16 in a heterologous exon context was responsive to PKA repression,Citation32 but this repression by PKA probably does not play a major role in PC12 cells since the PKA pathway does not appear required for the repression in these cells (). Tests with PKA using a reporter containing a 100 times longer CaMKK2 sequence (720 bp insert, ) indicates that the PKA effect on exon 16 is overall enhancing, consistent with the endogeneous PKA effect on exon 16 in B35 cells. For the opposite effects by PKA on the two mini-genes, a reasonable explanation is that the longer reporter contains PKA-responsive elements that enhance exon 16 inclusion and this effect overcomes the repressive effect through the CAA AAA A heptamer.
The opposite effect in the cell context-dependent splicing regulation is not alone. Similar regulation has also been observed for the stress axis-regulated exon (STREX) by depolarization in neurons and endocrine cells.Citation34,Citation35,Citation48 These observations on mini-gene and cell-dependent effects likely reflects the complex control of alternative exons by multiple signaling pathways as well as positive and negative elements/factors whose effects may vary between cells.Citation46,Citation47
Several neuron-specific splicing factors have been identified, including the Fox-1 and -2 proteins,Citation49 the Hu proteins,Citation50 the neuronal PTB (nPTB),Citation51–Citation53 as well as the Nova-1 and -2 proteins.Citation54 These factors are either enhancers or repressors of splicing dependent on the target exon/motif locations.Citation49,Citation50,Citation54–Citation56 For the exon E16, two copies of the Fox protein binding site UGCAUG are at the 196 nt and 219 nt positions in its downstream intron, where they are usually enhancers.Citation49 PTB target consensus motifs UUCU and CUCU were also seen in the pyrimidine-rich sequence of its upstream 3′ splice site. Nova target motifs UCAU and UCAC were also found in the upstream intron but not as clustered as in the known cases of regulated splicing.Citation54 The Hu protein target motifs A(U)3–5A,Citation57 were not found in/around E16. The significance of these and other factors in the forskolin/PKA control of CaMKK2 splicing requires further investigation.
In addition to its regulation by PKA ( and ), the alternative splicing of CaMKK2 is also controlled by membrane depolarization and CaMKIV.Citation32 Considering the presence of more than 500 kinase-encoding genes and the many splice variants of protein kinases exhibiting functional differences as mentioned above,Citation4–Citation18 it is possible that there are more alternative exons/splice variants in the kinome that are potential targets for similar cross- or feedback-regulation by other protein kinases or by a kinase itself. With extensive crosstalk or feedback regulation through alternative splicing in the signaling network, even a small shift in the splice variant composition would be expected to impact on various pathways and cellular processes. We thus propose that alternative splicing likely provides another level of regulation between protein kinases for a finely-tuned cell signaling network to orchestrate cellular processes.
Materials and Methods
Plasmid construction.
Wild type Flag-PKA and a kinase-dead mutant Flag-PKAm are as described previously in references Citation32 and Citation37. To make the CaMKK2 exon 16 splicing reporter minigene, a fragment containing the human exon 16 and partial flanking introns (353 bp upstream and 324 bp downstream) of the CaMKK2 gene was cloned between the Apa I and Bgl II sites of the pDUP175.Citation34 The control minigenes ANXA11 (Annexin A11), containing the exon 3 (as in NM_145869) and 25 bp upstream and 10 bp downstream partial flanking introns, and P2RX4 (purinergic receptor P2X4 isoform a), containing the exon 2 (as in CR609333) and 39 bp of upstream and 10 bp downstream partial flanking introns, were made similarly. To make pMyc-CaMKK2 −E16, a CaMKK2 cDNA (BC026060) fragment was amplified with primers rCaMKKb1S (sense strand, 5′-AGT CGG ATC CCA TCA TGT GTC TCT AGC CAG-3′, with BamH I site underlined) and rCaMKKb1A (antisense strand, 5′-AGT CGG TAC CCT ACT CcG GCT CCA TGG CCT CCT C-3′, with Kpn I site underlined. The lowercased rat-specific nucleotide does not change the corresponding amino acid proline) from a human CaMKK2 full length cDNA clone (Open Biosystems, Catalog No. MHS1011-7508895), digested with BamH I and Kpn I, and subcloned between the Bgl II and Kpn I sites of pCMV-Myc (Clontech). The original Bgl II site of the vector is lost upon ligation with the complementary BamH I end. For pMyc-CaMKK2 +E16, a 3′ end cDNA fragment containing exon 16 was amplified from a human neuronal cell line LAN-5,Citation58 with sense primer hCaMKK2BglII (5′-CTC TGA GAC CCG CAA GAT CTT C-3′, with BglII site underlined) and antisense primer rCaMKKb1A, digested with Bgl II and Kpn I and inserted between the same sites of pMyc-CaMKK2 −E16. Both CaMKK2 clones contain exon 14 sequence. The plasmid vector pGIPz for microRNA-adapted short hairpin RNA (shRNA) expression is a generous gift of Dr. Sam Kung. A clone for shRNA (shKK2) targeting a common region (nucleotides 701–719, in NM_031338) of the rat CaMKK2 sequence (5′-CAA TGA AAG TGC TGT CCA A-3′) was purchased from the Open Biosystems library (Cat# RMM4431-98978481) in the Proteomic Centre of the University of Manitoba.
2′-O-methyl RNA oligonucleotides.
Two oligonucleotides complementary to the splice sites of the exon 16 of CaMKK2: 5′-GUU UUU UGc ugg aau ugc cag-3′ (for 3′ splice site, nucleotides complementary to exon regions in upper and introns in lower cases) and 5′-agc agg cau uac CUU GGG CUC-3′ (for 5′ splice site), and one derived from the human ataxia-telegiectasis mutated (ATM) gene exon 43 (5′-CAA UGG UCC CCC UGC AUA UUC-3′, NM_000051) with no perfect match to rat sequences, were synthesized (by Integrated DNA Technologies), with the 2′-OH positions of the sugar backbones methylated for higher stability.Citation59 A short motif “aguguu,” shown to be effective in RNA localization in the nucleus,Citation60 was added to the 3′ end of these oligos.
Cell culture, transfection and treatment.
Rat neuroblastoma B35 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin-glutamine solution (Invitrogen) at 37° C in 5% atmospheric CO2.
Transient plasmid transfection using Lipofectamine 2000 (Invitrogen) was according to the manufacturer's instructions. The transfected B35 cells were cultured for overnight (MycCaMKK2 or shKK2) at 37° C, before treatment with forskolin (10 µM) for about 7 h followed by immunostaining. HEK293T cells were cultured and transfected as described in reference Citation32. 2′-OME-RNA oligos were transfected using OligoFectamine (Invitrogen). The cells were transfected for 2 days before treatment with forskolin or ethanol for 7 h, then fixed, treated with Triton-X 100 and incubated with phalloidin-TRITC plus DAPI.
For analysis of the alternative splicing of CaMKK2, B35 cells were plated at about 70% confluence in a six well plate, cultured for overnight and treated with either ethanol (vehicle) or forskolin (10 µM) for 6 h before RNA extraction. H89 (10 µM) was applied 15 min prior to forskolin addition. For immunostaining of the endogenous CaMKK2, B35 cells were treated with forskolin (15 µM) for 12 h.
RT-PCR.
This procedure was as described previously in reference Citation36. PCR reaction was in a 12.5 ul reaction for 25 cycles at an annealing temperature of 60° C, with 1.0 ul of RT product from ∼20 ng total RNA as templates. For CaMKK2 variants, PCR was primed by CaMKK2E14S (5′-ACC CGG ATG TTG GAC AAG AAC-3′) or CaMKK2S (5′-GAT CCT AGT GAA GAC CAT G-3′) together with γ-32P-ATP-labeled rCaMKK2A (5′-CCA CCT TTC ACA AGA GCA C-3′). For the minigene products, PCR was primed with Dup8a (5′-CTC AAA CAG ACA CCA TGC ATG G-3′) and γ-32P-ATP-labeled Dup10 (5′-CAA AGG ACT CAA AGA ACC TCT G-3′). The PCR products were separated in 4% or 6% denaturing PAGE gels and exposed to phosphorimager plates. The band intensities were quantified using the NIH ImageJ 1.41o software. The intensity percentage of the CaMKK2 containing exon 16 products was calculated.
In vitro protein kinase assay.
This procedure was adapted from that described previously in reference Citation38. Myc-CaMKK2 variants were expressed in HEK293T cells and immunoprecipitated with 2 µg of anti-cMyc (A14, Santa Cruz Biotechnology) as described in reference Citation33. The pelleted beads were then stored in DG buffer.Citation33 Both variants autophosphorylated linearly within a range of 30 ng to 120 ng in preliminary experiments, indicative of intramolecular phosphorylation as suggested.Citation45 Therefore, about 75–90 ng of the variant proteins were used in the following kinase assays. For autophosphorylation, Myc-CaMKK2 variants were added to 10 µl of kinase buffer (200 mM of Tris-Cl, 0.5 mM of DTT, 0.1% of Tween-20, 8 mM of MgCl2, 0.1 mM of ATP) with 2.5 µCi of γ-32P-ATP in the presence of 1 mM CaCl2 plus 1 µM of Calmodulin (CaM) or 1 mM of EGTA. For CaMKK2 kinase activity assay, 50 ng of GST-CaMKIV (BioMol, Cat#SE-377) were incubated with each Myc-CaMKK2 variant in the same kinase buffer. For PKA phosphorylation assay, 10 ng of PKA (Calbiochem, Cat#539486) and each Myc-CaMKK2 variant were incubated in the kinase buffer with 1 mM of EGTA. The reactions were incubated at 30°C for 20 min, terminated by boiling in 1x Lammli buffer and resolved in 4–20% SDS-PAGE gel. The gels were then dried, exposed to phosphorimager plates, scanned with a Bio-Rad phosphorimage scanner and finally subjected to western blots using antibodies as specified in the figure legend.
Immunostaining.
Immunostaining of B35 cells were adapted from that described in reference Citation33, using mouse anti-CaMKKβ (CaMKK2, ZZ9, Santa Cruz Biotechnology, 1:50) or rabbit anti-cMyc (A14, 1:100) as primary antibodies and a fluorescein-conjugated secondary antibody (green, 1:1,000). DAPI (blue, 1:6,000) and Phalloidin-TRITC (red, 1:3,000) were used to highlight the nuclei and F-actin fibers, respectively. The stained cells were kept in mounting media (Invitrogen, Cat#P36934) and observed under a fluorescent microscope. Images of cells stained with fluorescein, DAPI and Phalloidin-TRITC were captured through filters with emission wave lengths of 536/40, 447/60 and 624/40 nm, respectively.
Dual luciferase assay.
200 ng of the CRE reporter (SABioscience, Cat#CCS-002L) were co-transfected into B35 cells with 400 ng of pCMV-Myc, pMyc-CaMKK2 −E16 or pMyc-CaMKK2 +E16, respectively, for 38 to 46 hours. For luciferase activity measurement, cell lysates were prepared using Promega's Dual-Luciferase Reporter Assay system (Cat#E1910) according to the manufacturer's protocol, followed by reading on an EG&G Berthold LB96V microplate luminometer with a measure delay time of 1.6 seconds.
In vivo 32P-orthophosphoric acid labeling and phosphopeptide mapping.
This was carried out according to the procedure described previously in reference Citation37. Briefly, Myc-tagged CaMKK2 expression plasmids were each cotransfected with Flag-PKA or Flag-PKAm into HEK293T cells and incubated with 32P-orthophosphoric acid for 4 hours after fasting in Krebs-Ringer glucose buffer. The labeled Myc-CaMKK2 proteins were immunoprecipitated with anti-Myc antibody, resolved in 4–20% PAGE gels, blotted to nitrocellulose membranes, subject to chymo-trypsin and trypsin digestion and separation in a Hunter Unit followed by thin-layer chromatography.
Western blot.
Western blot was carried out as reported in reference Citation36, using mouse anti-cMyc (9E10, Clontech, 1:1,000) for the Myc-CaMKK2 variants and mouse anti-β-actin (C4, Santa Cruz biotechnology, 1:500) as primary antibodies.
For western blots of proteins in kinase assays, the dried gels were rehydrated with TBS buffer for 2 h and transferred onto polyvinylidene difluoride membranes (Millipore, Cat#IPVH00010). The blots were then probed with anti-cMyc or anti-CaMKIV (Cell Signaling Biotechnology, Cat#4032, 1:1,000) as primary antibodies.
Neurite length measurement and neurite branch counting.
Images of the anti-cMyc- or phalloidin-TRITC-stained or EGFP-expressing B35 cells were randomly captured and adjusted to high contrast for the measurement of neurite length. The longest neurite from each cell was measured with the Image-Pro MDA 6.0 software (Media Cybernetics, Inc.). The lengths of the H89 experiment, carried out at a later time, were normalized to the earlier ones. The numbers of primary and higher order neurites were counted manually. Statistical analyses were according to two-tailed Student's t-test.
Figures and Tables
Figure 1 Alternative splicing of the rat CaMKK2 gene. Diagram of the alternative splicing of rat CaMKK2 and the encoded variants. Exons (boxes) are numbered and their splicing patterns indicated with joined lines between exons. Arrowheads: primers used for RT-PCR of the endogeneous transcripts. *: the rat exon 17 corresponds to the exon 18 in human CaMKK2 named by Hsu et al. in JBC'97, 276:31113; rat exon corresponding to the human exon 17 were not found in the rat genome using BLAST searches, thus the rat protein products here corresponding to the human CaMKKβ1. Exon 16-encoded peptide sequences from rat and three other mammalian species are shown with unmatched amino acids highlighted. Among the protein domains, the exon 16-encoded region (hatched lines) is next to the CBD (Calcium-calmodulin-binding domain) and AID (autoinhibitory domain). The amino acid sequences of the resulting -COOH terminals of the rat CaMKK2 variants are aligned at the bottom, with the exon 16-encoded peptide underlined. Inclusion of the 43 nt exon 16 causes a frame shift leading to a longer -COOH terminal.
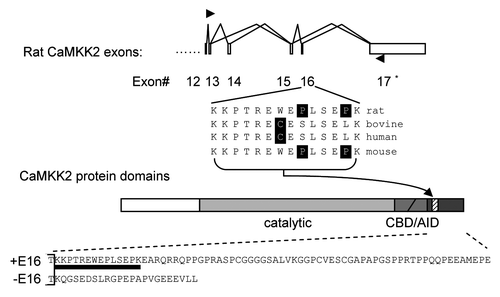
Figure 2 Cell-dependent effect of forskolin on exon 16 inclusion and the essential role of the PKA pathway in B35 cells. (A) A bar graph of the level of exon 16 inclusion upon treatment by forskolin (10 µM) with kinase inhibitors H89, PP1 and PP2 or their vehicle DMSO in PC12 cells. The level in the ethanol (ETOH)-treated samples is taken as 1.0. *p < 0.05. (B) cpt-cAMP and forskolin-induced PKA pathway increases the percentages of exon 16-included CaMKK2 transcripts (exon 16-containing band intensity/total product intensity) in B35 cells. Shown is a denaturing PAGE gel of spliced CaMKK2 variants in B35 cells non-treated (NT) or treated for 6 hours with either ethanol (ETOH), cpt-cAMP (100 µM) or forskolin (10 µM), with or without pretreatment with H89 (10 µM), as indicated above the gel. Indexes of exon 16 inclusion (relative to the NT sample level) are graphed under each lane (mean ± SEM, n = 17 except for forskolin, which is 10). Both of the CaMKK2 products contain exon 14, comprising the majority of transcripts (∼72%) encoding active CaMKK2 kinase isoforms in B35 cells. The kinase-dead exon 14-excluded variants are not shown here. **p < 0.01, ***p < 0.001. Nc: PCR negative control. (C) Effects of coexpressed Flag-PKA on splicing reporter minigenes in HEK293T cells. Upper, diagram of the splicing reporter minigenes containing CaMKK2 exon 16, ANXA11 exon 3 (Control 1) or P2RX4 exon 2 (Control 2). Boxes: exons; horizontal lines: introns; arrowheads: primers. Lower, PAGE gels of the RT-PCR products of the splicing reporters with coexpressed mutant (Mut) or wild-type (WT) Flag-PKA, with mean percentages of exon inclusion under each lane and exon-included or -excluded products indicated to the right. Lane 1 is a negative control of RT-PCR without reverse transcriptase. (D) A bar graph of the exon inclusion levels (mean ± SD) of the reporter minigenes and the PKA effect. The level in the PKA mutant (Mut)-coexpressing samples were taken as 1 for each reporter.
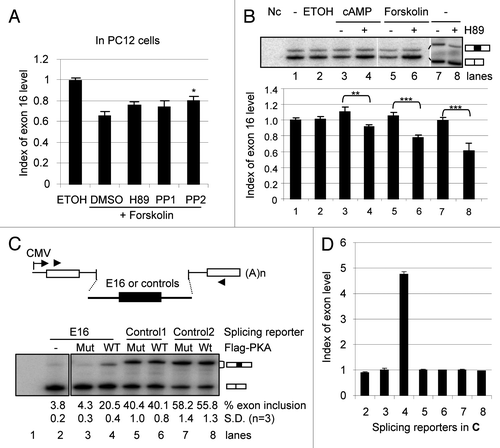
Figure 3 Biochemical properties of the Myc-CaMKK2 variants. (A) Upper, diagram of the Myc-CaMKK2 variants (domains are as in ). Lower, a western blot of Myc-CaMKK2 expressed in HE K293T cells probed with the anti-cMyc primary antibody. Both cDNAs contain exon 14 therefore encoding the kinase active isoforms. (B–D) Upper, In vitro kinase assays for the autophosphorylation (B) and phosphorylation of GST-CaMKIV (C) by Myc-CaMKK2 variants in the presence or absence of Ca2+ and Calmodulin (Ca-CaM), as indicated, as well as their phosphorylation by PKA (D). Phosphorylated Myc-CaMKK2 variants, PKA and GST-CaMKIV are indicated to the right of the gels. Lower, western blots with anti-cMyc or anti-CaMKIV antibodies accordingly, to show the protein input, as indicated. The level of the phosphorylation signals of the variants are indicated below the gels. (E) Phosphopeptide mapping of Myc-CaMKK2 −E16 and Myc-CaMKK2 +E16 immunoprecipitated from HE K293T cells with coexpressed wild type Flag-PKA. Left: immunoprecipitated 32P-labeled variant poteins (arrow head) used for peptide mapping. Spots consistently showed up for both variants are numbered to their left (#1–4) or right (#5–8). The intensities of spots #1 and 4 appear different here but not consistent between experiments, likely due to partial digestion. The Myc-CaMKK2 +E16-specific spot is circled. Orientations of the first (1°) and the second (2°) dimensions of the thin layer chromatography (TLC) plates are as indicated. (F) Transcriptional activities of Myc-CaMKK2 variants on a CRE (cAMP/calcium-responsive element) reporter in B35 cells in a dual-luciferase assay. Relative luciferase activities (mean ± SEM, n = 6) of either Myc-CaMKK2 −E16 (−E16) or Myc-CaMKK2 +E16 (+E16) were normalized to that of the vector control.
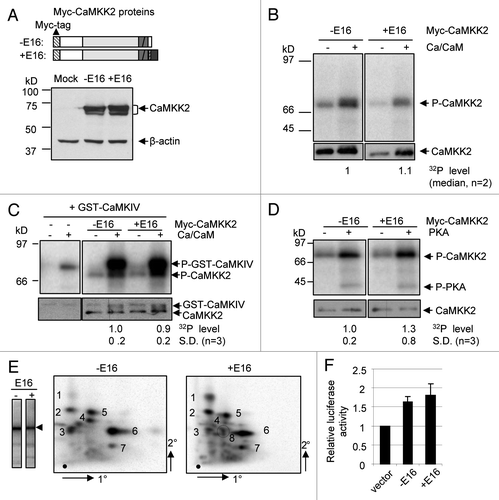
Figure 4 The endogenous CaMKK2 protein is localized in neurites and is required for forskolin-induced neurite growth in B35 cells. (A) B35 cells were treated with ethanol or 15 µM of forskolin for 12 h, fixed and incubated with Phalloidin-TRITC (left part), anti-CaMKKβ (CaMKK2, middle part) or DAPI (right part), and images collected through the respective filters as detailed in Methods. Magnification: 20x objective. (B) Distribution of CaMKK2 protein in the neurites of B35 cells treated with forskolin for 12 h. Images of Phalloidin-TRITC (left) and anti-CaMKKβ-stained (middle) neurites are merged into one (right), in red and green, respectively. Magnification: 100x objective. Images of the cell body staining are in the box at the upper-left corner of each image. (C–E) Effect on forskolin-induced neurite growth by RNA interference of the expression of the endogeneous CaMKK2 gene. At the left of (C) are representative images of the cells transfected with the vector pGIPz(-GFP) or the CaMKK2 shRNA plasmid shKK2(-GFP), as indicated. The dotted line marks the boundary of the GFP positive cell. The white arrowheads point to the GFP expressing cells. At the right is a bar graph of the relative levels of the CaMKK2 immunostaining intensities in control non-GFP cells (taken as the base level 1.0, C1 and C2 for GIPz and shKK2 plasmids, respectively) and in GFP positive (GIPz and shKK2 transfected) cells as indicated (mean ± SEM, n = 8 and 10 pairs, respectively). In (D and E) are the neurite lengths and branches per cell (mean ± SEM), respectively.
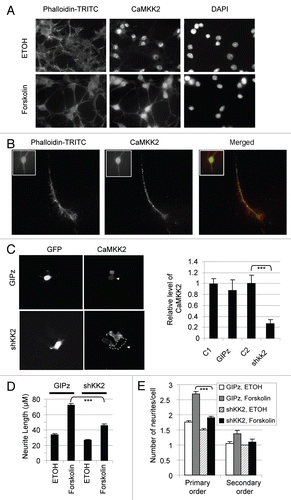
Figure 5 Myc-CaMKK2 variants differentially regulate forskolin-induced neurite growth of B35 cells. (A) Western blot of expressed Myc-CaMKK2 variants, with β-actin as a loading control. (B) Representative B35 cells expressing either Myc-CaMKK2 −E16 or Myc-CaMKK2 +E16, after forskolin (10 µM) or forskolin plus H89 treatment for 7 h. Shown are merged fluorescent images from anti-Myc antibody-immunostained (green) cells with the nuclei highlighted using DAPI (blue). (C) Mean (±SEM, n > 100, see text for details) neurite lengths of B35 cells expressing EGFP, Myc-CaMKK2 −E16 or Myc-CaMKK2 +E16. (D) Distribution of the neurite lengths (mean ± SEM) of the cells in (C). (E) Effects of Myc-CaMKK2 variants on neurite branching of the cells in (C). The bar graph shows the numbers (mean ± SEM) of primary, secondary and higher orders of neurites from each cell with the expression of EGFP, Myc-CaMKK2 −E16 or Myc-CaMKK2 +E16. Sample pairs with significant differences are marked with asterisks above the bars. *p < 0.05, **p < 0.01, ***p < 0.001. (F–I) Reduction of exon 16 changes forskolin-induced neurite growth. In (F) is a representative denaturing PAGE gel of RT-PCR products of B35 cells transfected with or without (NT) the 2′-OME oligos against the downstream 5′ splice site of exon 16 (E16) or a control exon. The oligo Control is derived from the ATM gene as detailed in the Methods. -: PCR negative control. In (G) are representative images of the transfected cells with ethanol or forskolin treatment. The far right column are larger views of the E16 oligo samples with several cells. In (H and I) are the bar graphs of the neurite lengths and number of branches per cell (mean ± SEM), respectively.
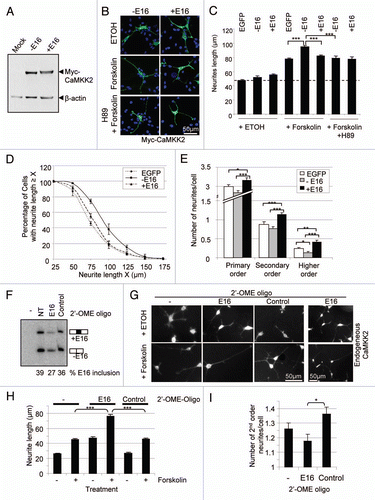
Figure 6 Brain-specific enrichment of the exon-16-containing transcripts. Shown are 32P-labeled RT-PCR products from an 8% denaturing PAGE gel, from RNA samples of different tissues as indicated above each lane. To the left are the molecular marker (M) sizes in nucleotides (nt). To the right are the expected product sizes and combination of exons 14 and 16 in each splice variant. -: PCR negative control lane for these samples. Beta-actin levels serve as RNA loading control and are shown below each lane. A bar graph of the level of CaMKK2 relative to beta-actin products from each lane is shown below the gels.
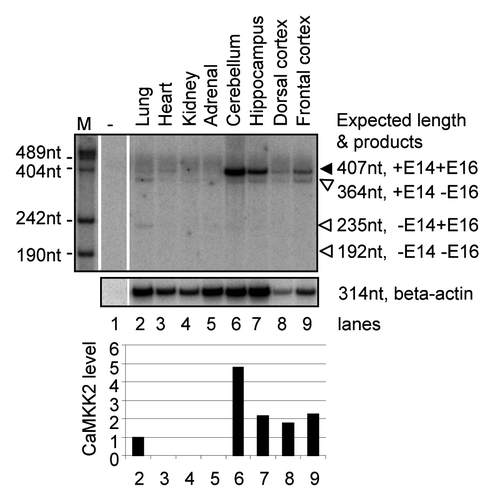
Figure 7 Summary for the splicing regulation of CaMKK2 by the PKA pathway. Stimulation of PKA by cpt-cAMP or forskolin increases the proportion of the CaMKK2 +E16 splice variant in B35 cells. The PKA effect on CaMKK2 exon 16 is inhibited by H89. The two CaMKK2 variants (black or gray ovals) are differentially phosphorylated by PKA. The CaMKK2 −E16 variant promotes the forskolin-induced neurite outgrowth while the CaMKK2 +E16 variant promotes neurite branching. ACI: adenylate cyclase type I.
Acknowledgments
We thank George Lawless, Allain Tobin, Sam Kung and Doug Black for cell lines and plasmids, and the labs of Jiming Kong and Aaron Marshall for valuable help. Supported to J.X. by the Canadian Institutes of Health Research (FRN68919 and FRN106608), the Manitoba Health Research Council (MHRC) and Canadian Breast Cancer Foundation Prairies/NWT. W.C. is a recipient of a postdoctoral fellowship and G.L. a graduate studentship from the MHRC, and J.X. a recipient of CFI infrastructure fund.
References
- Braconi Quintaje S, Orchard S. The annotation of both human and mouse kinomes in UniProtKB/Swiss-Prot: one small step in manual annotation, one giant leap for full comprehension of genomes. Mol Cell Proteomics 2008; 7:1409 - 1419
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298:1912 - 1934
- Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat Methods 2005; 2:17 - 25
- Ellenrieder C, Bartosch B, Lee GY, Murphy M, Sweeney C, Hergersberg M, et al. The long form of CDK2 arises via alternative splicing and forms an active protein kinase with cyclins A and E. DNA Cell Biol 2001; 20:413 - 423
- Kwon TK, Buchholz MA, Jun DY, Kim YH, Nordin AA. The differential catalytic activity of alternatively spliced cdk2alpha and cdk2beta in the G1/S transition and early S phase. Exp Cell Res 1998; 238:128 - 135
- Proszkowiec-Weglarz M, Richards MP, McMurtry JP. Molecular cloning, genomic organization and expression of three chicken 5′-AMP-activated protein kinase gamma subunit genes. Poult Sci 2006; 85:2031 - 2041
- Carling D, Aguan K, Woods A, Verhoeven AJ, Beri RK, Brennan CH, et al. Mammalian AMP-activated protein kinase is homologous to yeast and plant protein kinases involved in the regulation of carbon metabolism. J Biol Chem 1994; 269:11442 - 11448
- Verhoeven AJ, Woods A, Brennan CH, Hawley SA, Hardie DG, Scott J, et al. The AMP-activated protein kinase gene is highly expressed in rat skeletal muscle. Alternative splicing and tissue distribution of the mRNA. Eur J Biochem 1995; 228:236 - 243
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 1996; 15:2760 - 2770
- Coussens L, Rhee L, Parker PJ, Ullrich A. Alternative splicing increases the diversity of the human protein kinase C family. DNA 1987; 6:389 - 394
- Blobe GC, Khan WA, Halpern AE, Obeid LM, Hannun YA. Selective regulation of expression of protein kinase Cbeta isoenzymes occurs via alternative splicing. J Biol Chem 1993; 268:10627 - 10635
- Kawakami T, Kawakami Y, Kitaura J. Protein kinase Cbeta (PKCbeta): normal functions and diseases. J Biochem 2002; 132:677 - 682
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 2005; 120:59 - 72
- Bayer KU, De Koninck P, Schulman H. Alternative splicing modulates the frequency-dependent response of CaMKII to Ca(2+) oscillations. EMBO J 2002; 21:3590 - 3597
- Singh P, Leddy JJ, Chatzis GJ, Salih M, Tuana BS. Alternative splicing generates a CaM kinase IIbeta isoform in myocardium that targets the sarcoplasmic reticulum through a putative alphaKAP and regulates GAPDH. Mol Cell Biochem 2005; 270:215 - 221
- O'Leary H, Lasda E, Bayer KU. CaMKIIbeta association with the actin cytoskeleton is regulated by alternative splicing. Mol Biol Cell 2006; 17:4656 - 4665
- Shaw RJ. LKB1: cancer, polarity, metabolism and now fertility. Biochem J 2008; 416:1 - 3
- Towler MC, Fogarty S, Hawley SA, Pan DA, Martin DM, Morrice NA, et al. A novel short splice variant of the tumour suppressor LKB1 is required for spermiogenesis. Biochem J 2008; 416:1 - 14
- Xie J. Control of alternative pre-mRNA splicing by Ca(++) signals. Biochim Biophys Acta-Gene Regulatory Mechanisms 2008; 1779:438 - 452
- Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002; 420:691 - 695
- Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem 2008; 283:1223 - 1227
- Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev 2009; 23:482 - 495
- van der Houven, van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, Caceres JF. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. Journal of Cell Biology 2000; 149:307 - 316
- Shin C, Manley JL. Cell signaling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol 2004; 5:727 - 738
- Benderska N, Becker K, Girault JA, Becker CM, Andreadis A, Stamm S. DARPP-32 binds to tra2-beta1 and influences alternative splicing. Biochim Biophys Acta 2010; 1799:448 - 453
- Stojdl DF, Bell JC. SR protein kinases: the splice of life. Biochem Cell Biol 1999; 77:293 - 298
- Matsushita M, Nairn AC. Inhibition of the Ca2+/calmodulin-dependent protein kinase I cascade by cAMP-dependent protein kinase. J Biol Chem 1999; 274:10086 - 10093
- Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci 1999; 24:232 - 236
- Kitani T, Okuno S, Fujisawa H. Regulation of Ca(2+)/calmodulin-dependent protein kinase kinase alpha by cAMP-dependent protein kinase: II. Mutational analysis. J Biochem (Tokyo) 2001; 130:515 - 525
- Okuno S, Kitani T, Fujisawa H. Regulation of Ca(2+)/calmodulin-dependent protein kinase kinase alpha by cAMP-dependent protein kinase: I. Biochemical analysis. J Biochem (Tokyo) 2001; 130:503 - 513
- Wayman GA, Tokumitsu H, Soderling TR. Inhibitory cross-talk by cAMP kinase on the calmodulin-dependent protein kinase cascade. Journal of Biological Chemistry 1997; 272:16073 - 16076
- Li H, Liu G, Yu J, Cao W, Lobo VG, Xie J. In vivo selection of kinase-responsive RNA elements controlling alternative splicing. J Biol Chem 2009; 284:16191 - 16201
- Ma S, Liu G, Sun Y, Xie J. Relocalization of the polypyrimidine tract-binding protein during PKA-induced neurite growth. BBA-Mol Cell Res 2007; 1773:912 - 923
- Xie J, Black DL. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 2001; 410:936 - 939
- Xie J, Jan C, Stoilov P, Park J, Black DL. A consensus CaMK IV-responsive RNA sequence mediates regulation of alternative exons in neurons. RNA 2005; 11:1825 - 1834
- Yu J, Hai Y, Liu G, Fang T, Kung SK, Xie J. The heterogeneous nuclear ribonucleoprotein L is an essential component in the Ca++/calmodulin-dependent protein kinase IV-regulated alternative splicing through cytidineadenosine repeats. J Biol Chem 2009; 284:1505 - 1513
- Xie J, Lee JA, Kress TL, Mowry KL, Black DL. Protein kinase A phosphorylation modulates transport of the polypyrimidine tract-binding protein. Proc Natl Acad Sci USA 2003; 100:8776 - 8781
- Hsu LS, Chen GD, Lee LS, Chi CW, Cheng JF, Chen JY. Human Ca2+/calmodulin-dependent protein kinase kinase beta gene encodes multiple isoforms that display distinct kinase activity. J Biol Chem 2001; 276:31113 - 31123
- Nakamura Y, Okuno S, Kitani T, Otake K, Sato F, Fujisawa H. Immunohistochemical localization of Ca(2+)/calmodulin-dependent protein kinase kinase beta in the rat central nervous system. Neuroscience Research 2001; 39:175 - 188
- Sakagami H, Umemiya M, Saito S, Kondo H. Distinct immunohistochemical localization of two isoforms of Ca2+/calmodulin-dependent protein kinase kinases in the adult rat brain. Eur J Neurosci 2000; 12:89 - 99
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, et al. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci 2004; 24:3786 - 3794
- Weeks BS, Papadopoulos V, Dym M, Kleinman HK. cAMP promotes branching of laminin-induced neuronal processes. J Cell Physiol 1991; 147:62 - 67
- Francisco H, Kollins K, Varghis N, Vocadlo D, Vosseller K, Gallo G. O-GLcNAc post-translational modifications regulate the entry of neurons into an axon branching program. Dev Neurobiol 2009; 69:162 - 173
- Mercatante DR, Mohler JL, Kole R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J Biol Chem 2002; 277:49374 - 49382
- Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, et al. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. Journal of Biological Chemistry 1998; 273:31880 - 31889
- Hanamura A, Caceres JF, Mayeda A, Franza BR Jr, Krainer AR. Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA 1998; 4:430 - 444
- Smith CW, Valcarcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci 2000; 25:381 - 388
- An P, Grabowski PJ. Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol 2007; 5:36
- Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, Zhang MQ. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev 2008; 22:2550 - 2563
- Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci 2008; 65:3168 - 3181
- Markovtsov V, Nikolic JM, Goldman JA, Turck CW, Chou MY, Black DL. Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol Cell Biol 2000; 20:7463 - 7479
- Polydorides AD, Okano HJ, Yang YYL, Stefani G, Darnell RB. A brain-enriched polypyrimidine tractbinding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proceedings of the National Academy of Sciences of the United States of America 2000; 97:6350 - 6355
- Ashiya M, Grabowski PJ. A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: Evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA (New York) 1997; 3:996 - 1015
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, et al. An RNA map predicting Nova-dependent splicing regulation. Nature 2006; 444:580 - 586
- Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 2009; 36:996 - 1006
- Zhu H, Hasman RA, Barron VA, Luo G, Lou H. A nuclear function of Hu proteins as neuron-specific alternative RNA processing regulators. Mol Biol Cell 2006; 17:5105 - 5114
- Chung S, Jiang L, Cheng S, Furneaux H. Purification and properties of HuD, a neuronal RNA-binding protein. J Biol Chem 1996; 271:11518 - 11524
- Modafferi EF, Black DL. Combinatorial control of a neuron-specific exon. RNA (New York) 1999; 5:687 - 706
- Morvan F, Porumb H, Degols G, Lefebvre I, Pompon A, Sproat BS, et al. Comparative evaluation of seven oligonucleotide analogues as potential antisense agents. Journal Of Medicinal Chemistry 1993; 36:280 - 287
- Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science 2007; 315:97 - 100