Influenza A viruses activate the host phosphoinositide 3-kinase (PI3K) signaling pathway at several stages of their replication cycle, which depending upon the stage can have pro- or anti-viral consequences. For example, transient PI3K activation due to virion attachment appears to promote virus entry, while pathogen recognition receptor-mediated PI3K signaling may contribute to host innate immune defenses. In addition, the viral NS1 protein, a multifunctional virulence factor, directly binds and activates PI3K to enhance efficient virus replication. Here, we comment on the strain-specific viral requirement for NS1-activated PI3K in vitro and in vivo. Our observations with NS1 suggest that to clarify the complex interplay between PI3K signaling and viral activators, it is essential to understand the temporal and spatial patterns of kinase activation, as well as the specific PI3K subclass and enzyme isotypes involved. It is likely that each independent activation event regulates individual pathways with distinct biological outcomes.
Introduction
The last decade has witnessed a tremendous research effort to identify human host cell factors that regulate virus replication and virulence. Motivated by the need to find new antiviral targets for viruses that currently cause public health concerns (e.g., HIV, hepatitis C and influenza), virologists are reporting increasing numbers of virus-host protein:protein or protein:nucleic acid interactions, as well as multitudes of host genes and pathways that are important for determining the outcome of infection. These data have largely come from large-scale proteomics (e.g., yeast-2-hybrid analyses or affinity purifications combined with mass spectrometry), transcriptomics and genome-wide siRNA screens. The hope is that by finding ways to manipulate these virus-host interactions, novel intervention strategies can be identified. However, the sheer amount of data generated, together with subtle experimental differences, and the intrinsic complexities of cellular signaling networks, create a challenge for virologists when trying to understand the biological relevance of such findings. One example of this in the influenza A virus field is the role of the phosphoinositide 3-kinase (PI3K) pathway, a critical signaling checkpoint that regulates diverse cellular processes, including growth, survival, translation, vesicle trafficking, membrane biology and immunity. Components of PI3K-regulated signaling consistently appear as critical factors required for the influenza A virus replication cycle in genome-wide siRNA screens. Functionally, there is growing evidence to implicate specific PI3K pathways in the endocytic uptake of influenza viruses. Furthermore, several viral products have been proposed to directly or indirectly modulate the levels of PI3K activity during infection, which confusingly appear to have either pro- or anti-viral consequences. In this News and Views article, we briefly review the complexities of the PI3K signaling pathway and the different ways that influenza viruses interact with this important cellular regulator. We focus on our recent study regarding direct activation of PI3K signaling by the influenza A virus NS1 protein, a multifunctional virulence factor. Our data have lead us to hypothesize that for NS1-mediated PI3K signaling to have a pro-viral function, activation must occur at a distinct sub-cellular location and involve a specific PI3K regulatory isoform. We discuss the implications of these findings for our understanding of all PI3K signaling events during influenza A virus infection. We propose that spatial and temporal kinetics intersect with isoform-dependent signaling to delineate separate biological outcomes for each PI3K activation event.
Class IA PI3K: Signaling Inputs and Outputs
Phosphoinositide 3-kinases (PI3K) are a family of cellular enzymes that phosphorylate the 3-hydroxyl group on the inositol ring of membrane-embedded phosphatidylinositol lipids, thereby generating second messenger molecules that regulate the intracellular distribution and activity of various cell signaling proteins. Biological processes regulated in this way broadly include cell growth, survival, proliferation, intracellular trafficking, cytoskeletal rearrangements and migration. Class I PI3K comprises obligate heterodimeric protein complexes consisting of a catalytic subunit (termed p110) and a regulatory subunit (p85-type, p101 or p87), and use phosphatidylinositol (4,5) bisphosphate [PtdIns(4,5)P2] as their main substrate. Subclass IA PI3K includes those catalytic subunit isotypes (p110α, p110β and p110δ) that bind the p85-type (p85, p55 and p50) regulatory subunits. As each of the two main p85-type regulatory subunits (p85α and p85β) can heterodimerize with any of the three p110 catalytic subunits, there are at least six types of p85-mediated class IA PI3K heterodimer. Nevertheless, each heterodimer utilizes the same substrate in the same enzymatic reaction to produce the same second messenger product. Mechanistically, it is unclear how this same reaction output can be translated into biologically distinct outcomes, but given the conservation of different p85 and p110 isotypes and data from genetic ablation studies in mice, it is obvious that functional output differentiation must exist.
Canonically, in the absence of a stimulus, PI3K p110-p85 heterodimers remain inactive in the cytoplasm: the default function of the regulatory p85 subunit is to inhibit the catalytic p110 subunit via specific intermolecular contacts. Upon various stimulatory signaling inputs, including external stimuli such as growth factors or cytokines, the inactive complex moves to the plasma membrane, where the SH2 domains of p85 dock with phosphotyrosine residues present in receptor-associated signal-transduction proteins (summarized in ). This leads to a rearrangement of the intermolecular interactions between p85 and p110, such that the enzymatic activity of p110 is no longer inhibited, thereby stimulating production of the intracellular second messenger, phosphatidylinositol (3,4,5) trisphosphate [PtdIns(3,4,5)P3], from PtdIns(4,5)P2. Downstream of PI3K activation, several effector proteins that possess pleckstrin homology (PH) domains [or other PtdIns(3,4,5)P3-regulated domains] are recruited to the newly generated PtdIns(3,4,5)P3, and nucleate a plethora of different signaling cascades.
Figure 1. Physiological activation of class IA PI3K. The p110-p85 heterodimers, cytoplasmic in their basal state, dock to cellular membranes upon activation of various signal receptors, including receptor tyrosine kinases. A conformational change in p85 relieves the p110 catalytic site from inhibition by the p85 SH2 domains, and PtdIns(3,4,5)P3 is generated. Akt (and others with pleckstin homology (PH) domains) bind to PtdIns(3,4,5)P3 and build up local signaling platforms for multiple cellular pathways. SH3, Src-homology 3; BH, breakpoint-cluster region homology; nSH2, N-terminal Src-homology 2; iSH2, inter-SH2; cSH2, C-terminal Src-homology 2.
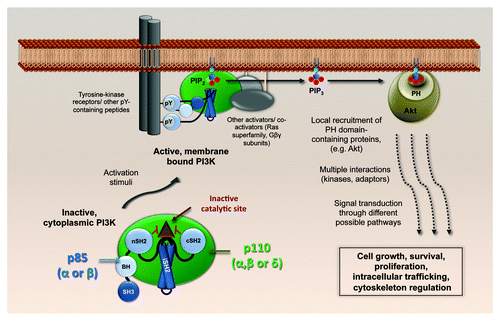
The Ser/Thr kinase Akt is the best-studied PH domain-containing effector downstream of PI3K, and its activating phosphorylation at Ser-473 following PtdIns(3,4,5)P3 binding has been used as a universal cell-based reporter for PI3K activity. However, several other mechanisms exist to increase PI3K versatility. The variety of PH domain-containing proteins besides Akt greatly expands the range of signals that can be propagated from a single second messenger such as PtdIns(3,4,5)P3. As stated above, the conservation of different p85 and p110 isotypes must lead to distinct activities, although these are poorly understood. Nevertheless, there is some evidence to suggest a p85-specific bias in directing the PI3K heterodimer toward distinct interaction partners, such as certain growth factor receptors that have been activated by specific ligands. Moreover, although PtdIns(3,4,5)P3 is mainly generated at the plasma membrane, it can also be found on nuclear and endocytic membrane compartments. Thus, the intracellular distribution of PtdIns(3,4,5)P3 pools could influence effector protein composition at particular sites, thereby differentially regulating functional propagation of PI3K signaling. Furthermore, in addition to their lipid kinase activity, several p110 subunits have protein kinase activity that may be specific to certain cellular substrates, and there have been proposals that some p110 subunits have distinct non-catalytic, scaffold-like activities. Direct activation of small GTPases (Ras, Rac and Arf superfamilies) by PI3K has also been described. In summary, the cellular PI3K system is a highly regulated system that can discriminate inputs from a variety of extracellular stimuli and convert them into specific functional outputs. The multitude of PI3K isotypes, their intracellular locations and their differential protein/lipid kinase or non-catalytic activities likely contribute to this.
PI3K Activation during Influenza A Virus Infection
Influenza A viruses are the etiological agents of an acute respiratory disease in humans. These viruses cause both seasonal outbreaks and occasional pandemics, and remain a major global health concern despite our improving knowledge on their biology, and the development of intervention strategies such as vaccination. Influenza A viruses encode 10–12 polypeptides on their 8-segmented, negative-sense RNA genome. Given this small coding capacity, the virus is highly dependent upon, and must interact extensively with, various host-cell signaling components to influence both the viral replication cycle and disease outcome. As described below, the PI3K signaling pathway is an intriguing example of this, as influenza A viruses appear to encode several factors that stimulate activation of this pathway.
Initial studies on the effect of influenza A virus infection on PI3K signaling (by monitoring Akt Ser-473 phosphorylation levels) detected two temporally regulated waves of activation. The first wave appears to be weak, transient, and occurs very early during infection (~15–30 min post-infection). Some studies have proposed that influenza virus particle attachment to sialic acid-containing receptors on the surface of cells it is entering triggers the clustering of receptor tyrosine kinases, thereby mimicking canonical activation of class IA PI3K by growth factors. This, in turn, may promote endocytosis and contribute to acidification of late endosomes by activating v-ATPases, a critical step in triggering virus-host membrane fusion and the release of incoming viral genomes into target cells. Furthermore, Ras-mediated activation of another PI3K class (IB) has also been suggested to potentiate clathrin- and Akt-independent endocytosis of influenza viruses.
The second wave of PI3K activation during influenza A virus infection occurs at intermediate-to-late time points (~4–6 h onwards), is characterized by high levels of phosphorylated Akt and is mediated by expression of the viral NS1 protein, a multifunctional virulence factor with well-defined roles in antagonizing the host innate immune system. NS1 specifically and directly binds the p85β regulatory subunit of PI3K, but is unable to bind p85α, an intriguing observation for which the reasons are unknown. In vitro assays have subsequently indicated that such an interaction forms the core of a heterotrimeric complex between NS1, p85β and an inhibition-relieved, catalytically active p110 (). Structural, functional, and biochemical studies have characterized the interacting residues in both NS1 and p85β. Among them, the highly conserved tyrosine at position 89 of NS1 (Y89) is critically positioned at the interface between NS1 and p85β. Conservative substitution of tyrosine 89 to phenylalanine (Y89F) abrogates the NS1-p85β interaction, inhibits PI3K/Akt activation and attenuates virus growth in tissue culture. Perhaps unsurprisingly, given the underlying complexity of the PI3K network and its multiple functional outputs, addressing the biological relevance of PI3K activation by NS1 has been a challenging question. Early work suggested a link between NS1-mediated PI3K activation and a delay in virus-induced apoptosis, which is consistent with a clearly established anti-apoptotic role for the PI3K/Akt pathway. However, other studies have provided data to indicate that even though NS1 has anti-apoptotic properties, these are independent of PI3K activation, at least in the context of the influenza virus strains tested so far. Another study, more focused on tissue-specific activities, indicates that NS1-activated PI3K can play a role in regulating cation currents (and thereby liquid accumulation) in the respiratory epithelium of the lung. Many factors may account for these discrepancies, not least experimental variation in the viral strains and cell types used, the NS1 mutations that abrogated p85β binding, and the use of pharmacological inhibitors with unknown specificities and off-target effects.
Figure 2. A model of NS1-activated PI3K. NS1 binds the iSH2 domain of p85β and displaces the inhibitory nSH2 domain. The docking of NS1-activated PI3K complexes to membranes, the involvement of effectors other than Akt and the ultimate biological consequences remain unclear. Different viral strains and PI3K isotypes may drive the response through distinct pathways.
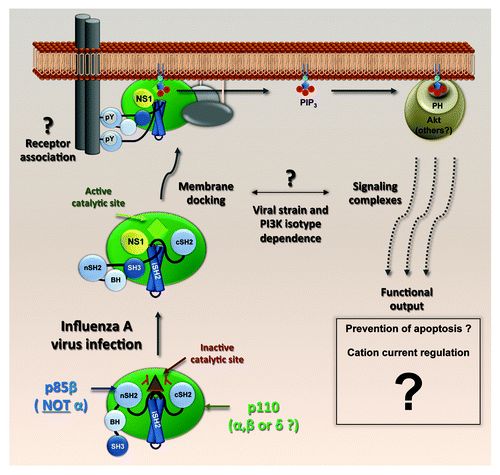
Although NS1 appears to be the main activator of PI3K during influenza A virus infection, other viral products also stimulate this signaling pathway, thereby adding layers of complexity. In particular, viral genomic RNA (vRNA) may trigger a PI3K-dependent antiviral response leading to production of interferon. Such vRNA-stimulated PI3K signaling seems to be mediated by the host viral RNA sensor, RIG-I, and should have a biological effect (e.g., anti-viral) opposite to that of NS1-activated PI3K (e.g., pro-viral). An unresolved question is therefore how absolute activation of PI3K by distinct viral inputs can lead to varying consequences for the virus at different stages of the replication cycle.
Strain-Specific Consequences of NS1-Activated PI3K: Not All PI3K Events are Equal?
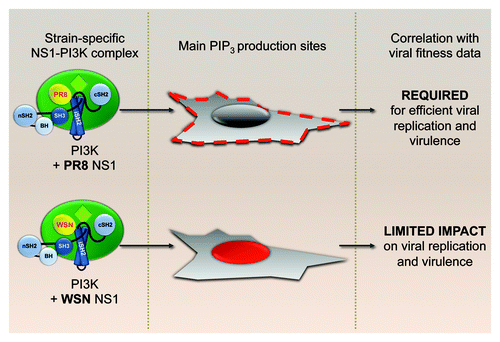
Previous studies had shown that introduction of the NS1-Y89F mutation into an H3N2 strain of influenza virus (A/Udorn/72) abrogates PI3K activation and attenuates virus replication during infection of tissue culture cells. We were initially interested in assessing the contribution of NS1-activated PI3K to influenza virus virulence in vivo, something that had not been previously assessed. For this purpose, we used the well-established mouse model of influenza A virus infection and introduced the NS1-Y89F mutation into two widely used mouse-lethal H1N1 strains of influenza virus: A/Puerto Rico/8/34 (PR8) and A/WSN/33 (WSN). In tissue culture, the mutant PR8 virus (rPR8 NS1-Y89F) was clearly attenuated as compared with its wild-type counterpart. However, there was no appreciable difference between the wild-type and mutant WSN viruses, despite clear differential activation of PI3K. Given the variety and cell type-specific activities of PI3K, we hypothesized that the effect of NS1-activated PI3K could be limited in the homogeneous context of tissue culture, but still have a significant impact within a complex animal model. Hence, it was surprising to find that the phenotypic difference between strains was maintained in vivo: mice infected with rPR8 NS1-Y89F showed less morbidity and mortality that those infected with wild-type rPR8, while no significant differences were observed between the wild-type and mutant WSN viruses. In accordance with this, mutant PR8 virus grew to lower titers in mouse lungs than wild-type, but there was no significant difference in lung titers between wild-type WSN and the NS1-Y89F mutant.
Both PR8 and WSN NS1 proteins are able to bind p85β and promote phosphorylation of Akt. Furthermore, it is clear that the Y89F mutation abrogates binding and activation of PI3K for both strains. Thus, it was intriguing to find that removal of the PI3K activating phenotype from these two strains lead to different functional consequences. Interestingly, we found a clear difference between PR8 and WSN in the intracellular localization of PtdIns(3,4,5)P3 (the catalytic product of activated PI3K) after co-expression of each NS1 protein with p85β and p110α. Cells transfected with PR8 NS1 showed increased levels of PtdIns(3,4,5)P3 accumulating in discrete domains of the plasma membrane that we have yet to identify, while WSN NS1-induced PtdIns(3,4,5)P3 preferentially accumulated in nuclear and perinuclear areas and was not detectably enriched at the plasma membrane. From these results, we postulated that the subcellular location of PI3K activation defines the functional output. In other words, although the initial players involved in the process remain the same [i.e., NS1, PI3K, PtdIns(3,4,5)P3 and Akt], the pathways they activate downstream (together with their respective biological consequences) depend on the spatial platform from which they are signaling. Thus, we hypothesize that the PI3K signaling cascade initiated at discrete domains of the plasma membrane has a different function to that initiated in the perinuclear/nuclear regions. Accordingly, the PR8 strain is sensitive to defects in activating this specific pathway, while the WSN strain is not (). Presumably, the WSN strain does not require plasma membrane-mediated NS1-activated PI3K for its replication strategy, and it is possible that the ability of this NS1 protein to activate PI3K is a remnant of viral requirement prior to laboratory and mouse adaptation. Multiple issues remain to be elucidated, including the molecular mechanism behind the strain-specific NS1 behavior (our data suggest that NS1 carries the information required to drive PI3K activation to a particular location), the identity of different platform-specific pathways activated and the reason for the apparent lack of biological relevance for the WSN NS1-induced perinuclear/nuclear accumulation of PtdIns(3,4,5)P3. At this point we cannot rule out that WSN NS1-activated PI3K may have biological consequences for the virus in other experimental systems beyond tissue culture cell-lines and murine models.
Figure 3. Strain-specific localization of PI3K activation. Viral strain specificity of PtdIns(3,4,5)P3 generation patterns by NS1-activated p110α-p85β complexes and their correlation with virus performance profiles in vitro and in vivo.
Widening the Scope beyond Location, Location, Location?
We believe that our observations on the strain-specific spatial activation of PI3K by NS1 and the assumed different signaling and biological consequences this has on virus replication are applicable to our understanding of other PI3K signaling events during influenza virus infection. Thus, future studies on PI3K activation should address spatial distribution during infection and focus on specific subcellular events rather than broad global reporters such as total Akt phosphorylation. In this way, we may begin to resolve the functional consequences of PI3K activity downstream of such diverse stimuli as virion binding, NS1 and vRNA-mediated activation of RIG-I. It may be that the other activation events also have strain-specific outcomes that need to be considered, and it will certainly be important to identify the kinetics of activation with each stimulus and combine this with the spatial data. For example, given that NS1 location varies over the course of infection, different NS1-PI3K signaling foci could be activated successively by the NS1 protein of a single viral strain, with each focus leading to a different biological outcome.
Another critical parameter that has yet to be taken into account regarding influenza A virus-mediated PI3K activation is the actual variety that exists within the generic term “PI3K.” An increasingly important area of research in the PI3K field is therefore to understand the different roles of various PI3K isotypes. In this regard, the functional consequences of NS1 specificity toward activating PI3K complexes that contain p85β, but not p85α, are not understood. Whether there is preferential, parallel, redundant or synergistic activation of the different catalytic p110 subunits by NS1 is also a pertinent question, especially given the tissue and cell specificity of these isotypes and the array of different cell types that influenza A viruses can infect in a host organism. This gap needs to be addressed not only for NS1, but also for the other viral activators of PI3K. Due to their clinical relevance in cancer and chronic inflammation, much effort has been put into developing better PI3K (usually p110) isotype-specific inhibitors and antibodies, many of which are now commercially available. Their use may help to clarify specific signaling activities and functional phenotypes associated with the diverse p110-p85 complexes.
Concluding Remarks
To fully understand how influenza A viruses interact with the host PI3K network, multiple variables must be considered. These include viral strain, host-cell type, time post-infection, subcellular location of the activation events and the PI3K isotypes involved. The consequences of successfully portraying a clear picture of this virus-host interaction could reach far beyond the virology field, given the relevance of PI3K for almost all physiological and pathological processes from development to oncogenesis. As viruses have been “learning” the intracellular molecular biology of our cells for a lot longer than academic biologists, the nuances and specificities of viral proteins such as the influenza A virus NS1 protein may become invaluable tools with which to probe cell signaling pathways.
Acknowledgments
We apologize to authors whose primary research papers were not cited due to the format of this News and Views article.