Upon infection of many bacterial pathogens, bacterial invasion is quickly sensed by the innate immune system, resulting in acute inflammatory responses. However, it is still unclear how pathogens modulate host inflammatory responses to bacterial internalization into epithelial cells. Here we provide evidence that the Shigella flexneri effector OspI delivered via the type III secretion system dampens acute inflammatory responses during bacterial invasion of epithelial cells by targeting TNF receptor-associated factor 6 (TRAF6). We found that OspI can bind to UBC13, an E2 ubiquitin-conjugating enzyme, and act as a unique glutamine deamidase by selectively deamidating Gln100 to Glu100 in UBC13. Consequently, the E2 ubiquitin-conjugating activity that is required for TRAF6 activation was inhibited, allowing S. flexneri OspI to modulate the diacylglycerol-CBM complex-TRAF6-NFκB signaling pathway. We determined the 2.0 Å crystal structure of OspI, which contains a putative Cys-His-Asp catalytic triad. A mutational analysis showed that this catalytic triad was essential for the deamidation activity. Our results highlight a unique bacterial tactic that modulates acute inflammatory responses to bacterial invasion of epithelial cells by targeting the UBC13-TRAF6 complex.
Many bacterial pathogens can enter various host cells where they promote their intracellular survival, transiently evade humoral immunity and further disseminate into other cells and tissues. When bacteria enter host cells and replicate intracellularly, the cells use various pattern recognition receptors (PRRs) to sense the invading bacteria either through damage-associated molecular patterns (DAMPs) or various pathogen-associated molecular patterns (PAMPs). As a result, the host cells induce alarm signals that activate the innate immune system to target and eliminate these pathogens. For example, upon invasion of the intestinal epithelium, some bacterial pathogens, such as S. flexneri and Listeria monocytogenes, induce the disruption of vacuolar membranes to facilitate their entry into the host cytoplasm. This membrane rupture is quickly sensed as DAMPs, which subsequently trigger acute inflammatory responses and occasionally autophagy by activating various cell signaling pathways. In addition, after S. flexneri enters host cells, the bacteria multiply therein and then spread to neighboring cells, during which the bacteria release peptidoglycan, lipopolysaccharide and flagellin. These bacterial components are recognized as PAMPs by cytoplasmic PRRs, such as Nod-like receptors (NLRs), that induce NFκB-mediated and MAPK-mediated inflammatory responses. Therefore, bacterial pathogens deploy various mechanisms to modulate the host immune responses. Indeed, S. flexneri delivers several effectors that modulate inflammatory signaling, including IpaH9.8, OspF, OspG and OspZ. However, it is still unclear what mechanisms bacteria use to modulate host responses, especially early innate immune responses to bacterial invasion of epithelial cells in which vacuolar membrane rupture is detected as DAMPs. In order to gain further insight into these mechanisms, we searched for S. flexneri effectors that are delivered via the type III secretion system (T3SS) and modulate acute inflammatory responses to bacterial invasion of epithelial cells. In our current study, we identified OspI, which is an uncharacterized T3SS-secreted effector encoded by ORF169b on the large (230 kb) virulence plasmid of S. flexneri. We determined that OspI is a key player in dampening acute inflammatory responses to bacterial invasion of epithelial cells.
We initially performed a comprehensive microarray analysis of HeLa cells infected with YSH6000 (wild-type S. flexneri), ΔospI (ospI-deficient mutant) or S325 (T3SS-deficient mutant) in order to assess the role of OspI in infection. We found that the levels of various chemokines (e.g., IL-8, CCL20, CXCL2 and CCL2) and cytokines (TNF and IL-6) in ΔospI-infected cells were markedly increased at 60 min after infection. This elevated chemokine and cytokine production was detected as early as 30 min post-infection and persisted until 60 min and 120 min post-infection. These changes in chemokine and cytokine levels during ΔospI infection were also observed in Caco-2 cells, a human colonic carcinoma cell line. Since these chemokines and cytokines are induced via activation of the NFκB and MAPK pathways, we subsequently examined immune signaling in ΔospI-infected HeLa cells. As expected, increased phosphorylation of IκBα and JNK1/2 was detected in HeLa cells that were infected with ΔospI but not YSH6000. Importantly, increased IκBα phosphorylation was detected as early as 10 min after infection during the early stage of S. flexneri invasion of epithelial cells. At 20 min post-infection, NFκB (p65) nuclear translocation was 4-fold higher in cells infected with ΔospI than YSH6000. Based on these results, we reasoned that OspI acts as a novel effector that dampens acute inflammatory responses to S. flexneri invasion.
To establish the in vivo role of OspI, we exploited a guinea pig rectum infection model, which is the most reliable model to evaluate S. flexneri pathogenesis. Guinea pigs were intrarectally inoculated with 1 × 109 cfu of YSH6000 or ΔospI and then the intestines were examined at 24 h after intrarectal challenge. ΔospI-infected rectums had increased IL-8 mRNA levels compared with YSH6000-infected rectums. Importantly, there were fewer ΔospI bacteria than YSH6000 bacteria in the rectal tissue at 24 h after intrarectal challenge. ΔospI infection also resulted in severe inflammation in the rectal epithelium layer with monocytes infiltrating into the lamina propria and this phenotype was much more profound compared with YSH6000 infection, suggesting that OspI contributed to S. flexneri pathogenesis.
We subsequently determined whether OspI inhibits NFκB activation upon S. flexneri infection of HeLa cells and found that ectopic OspI expression further inhibited NFκB activation. Previous studies showed that S. flexneri infection of epithelial cells resulted in activation of the Nucleotide-binding oligomerization domain 1 (NOD1)-RIP2-dependent and NOD1-RIP2-independent NFκB signaling pathways. The NOD1-RIP2-dependent pathway can be stimulated via PAMPs, while the NOD1-RIP2-independent pathway is stimulated via DAMPs. Therefore, we investigated how OspI suppressed NFκB activation using HeLa cells transiently expressing OspI (HeLa/OspI) or a mock control. When HeLa/OspI cells were stimulated with TNFα, NOD1 or phorbol 12-myristate 13-acetate (PMA) and then examined for NFκB activation, it was determined that OspI suppressed PMA-mediated, but not TNFα- or Nod1-mediated, NFκB activation. Since PMA is a substitute for diacylglycerol (DAG) in the activation of the protein kinase Cs (PKCs)-NFκB pathway and DAG in the host membrane acts as an important cue to trigger antibacterial autophagy against Salmonella enterica serovar Typhimurium, we hypothesized that OspI may selectively target a DAG-dependent NFκB signaling pathway during S. flexneri invasion of epithelial cells. Therefore, we used confocal immunofluorescence microscopy to examine membrane ruffles protruding around S. flexneri entry sites in HeLa cells expressing PKC-C1-GFP (PKC-C1 region fused to GFP as a DAG sensor) and found that DAG accumulated around the bacterial entry site. Indeed, increased IL-8 production in ΔospI-infected cells was suppressed by treating with Propranolol, a DAG inhibitor. The DAG-NFκB pathway is mediated through the CARD (CARD9, 10, 11 and 14)-BCL10-MALT1 (CBM) complex in lymphoid, myeloid and non-myeloid cells. The CBM complex is a major regulator of NFκB signaling in both innate and adaptive immunity. Thus, we examined if OspI modulates CBM complex-mediated NFκB signaling by knocking down BCL10 levels with siRNA and found that IL-8 levels were greatly decreased compared with the control siRNA. Furthermore, we found that GFP-MALT1 was recruited to the S. flexneri entry point in HeLa/GFP-MALT1 cells, since MALT1 functionally interacts with BCL10. These results suggested that DAG that colocalized with S. flexneri-induced membranes stimulates the DAG-CBM complex-NFκB pathway and that OspI specifically dampens this pathway.
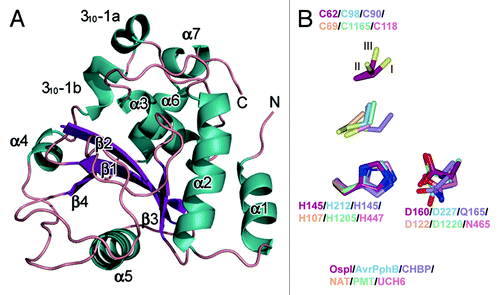
To gain further structural and functional insight, we determined the crystal structure of recombinant S. flexneri OspI at 2.0 Å resolution (PDB ID code 3B21). OspI had an α/β fold with four β-strands (β1–β4), seven α-helices (α1–α7) and one 310 helix. The structure was organized around a central anti-parallel β-sheet, with α-helices packed on both sides of the sheet (). A search of known structures in the Protein Data Bank revealed that OspI shared structural homology with a cysteine protease family and was most closely related to AvrPphB with a root mean square deviation (r.m.s.d.) value of 3.3 Å. AvrPphB is a Pseudomonas syringae T3SS effector and a member of a superfamily of related enzymes containing papain-like cysteine proteases, acetyl transferases, deamidases and transglutaminases. While there is considerable divergence in the overall fold across this superfamily, a core anti-parallel β-sheet and an α-helix containing the active site cysteine, which packs against the β-strands, are conserved across this family. A potential catalytic triad [cysteine (C, Cys) 62, histidine (H, His) 145 and asparagine (D, Asp) 160] in OspI was identified based on a comparison with the active site of AvrPphB. Superimposing His145 and Asp160 of OspI onto His212 and Asp227 of AvrPphB or other members of this superfamily revealed remarkable similarity (). However, Cys62 of OspI existed in three discrete conformations in the crystal structure, and the Sγ position was located on the opposite side of the active site in AvrPphB. The fractional occupancy of each conformer was estimated to be 0.55 (conformation A), 0.35 (conformation B) and 0.1 (conformation C). The highest occupancy site of Cys62 appeared to form a disulfide bond with Cys65 at 2.05 Å ().
Figure 1. Crystal structure of Shigella OspI. (A) Overall structure of Shigella OspI. The colors of the secondary structural elements indicate the following: dark green, α-helix; dark purple, β-strands; salmon, loops. (B) The alignments of the catalytic cores using the putative catalytic triad residues, all atoms of His, and the main chain atoms of Asp are shown as a reference. Dark red, Shigella OspI; light blue, AVRPphB (PDB ID code 1UKF); light purple, CHBP (Cif homolog from Burkholderia pseudomallei, PDB ID code 3GQM); orange, NAT (N-acetyltransferase, PDB ID code 1E2T); light green, PMT (Pasteurella multocida toxin, PDB ID code 2EBF); pink, UCH6 (ubiquitin C-terminal hydrolase 6, PDB ID code 1VJV). The C62 in OspI is represented in three alternate conformations with the three conformers labeled I, II and III.
To confirm that the C-H-D triad is the catalytic center within OspI, we substituted C62, H145 and D160 with serine (S) (for 62) or alanine (A) (for C62, H145 and D160), and these OspI mutants were examined for their ability to suppress NFκB activation. Complementing the ΔospI mutant with plasmids encoding the ospI (C62S), ospI (H145A) or ospI (D160A) genes did not reduce the increased IκBβ phosphorylation and IL-8 induction that were observed in ΔospI-infected HeLa cells. Consistent with this result, an NFκB reporter assay showed that OspI (C62S), OspI (H145A) and OspI (D160A) lost the ability to suppress NFκB activity upon S. flexneri infection or PMA stimulation. To investigate the possible involvement of Cys65 in OspI activity, we substituted Cys62 with serine (S) and then examined the effects of OspI C65S on IL-8 expression during S. flexneri infection. OspI Cys65 had no effect on OspI activity. Together these results indicated that C62, H145 and D160 in OspI are the catalytic triad that suppresses NFκB signaling.
We subsequently determined which steps in the DAG-CBM complex-NFκB pathway are targeted by OspI by examining NFκB activity in HeLa/OspI or HeLa/OspI (C62S) cells that were induced with BCL10, TRAF6, TAK1/TAB1, IKKβ or NFκB (p65). OspI but not OspI (C62S) suppressed NFκB activity when HeLa cells were induced with BCL10 and TRAF6 but not with TAK1/TAB1, IKKβ or p65, suggesting that OspI targets TRAF6 or an upstream step. Thus, we examined the possibility that OspI modulates TRAF6 activation during S. flexneri infection. We used rescued Traf6-deficient mouse embryo fibroblasts (MEFs) [Traf6−/−/WT-TRAF6, Traf6−/−/TRAF6 (C70A; E3 ligase-deficient mutant) and Traf6−/−/mock] and examined the effects of YSH6000 (wild-type S. flexneri) or ΔospI infection on Cxcl2 mRNA production. When Traf6-deficient MEFs were rescued with TRAF6 (C70A) or the mock control, the Cxcl2 levels induced upon ΔospI infection were greatly decreased compared with that rescued by wild-type TRAF6, suggesting that the ΔospI phenotype depends on TRAF6 E3 ligase activity. Based on these results, we concluded that OspI interferes with TRAF6 activation during S. flexneri infection.
TRAF6 is an E3 ubiquitin ligase that cooperates with ubiquitin (Ub)-activating E1 and Ub-conjugating E2 enzymes (UBC13 and UEV1A), which are required for TRAF6 self-ubiquitination and TRAF6-induced NFκB activation. Therefore, we investigated the effects of OspI and OspI (C62A) on TRAF6 using an in vitro self-ubiquitination assay and found that OspI, but not OspI (C62A), dampened TRAF6 polyubiquitination. However, OspI did not affect the formation of E2~Ub thioester intermediates, suggesting that OspI modifies TRAF6, UBC13, UEV1A or ubiquitin. We incubated OspI with each of these putative targets and examined their electrophoretic mobility using native PAGE and found that the mobility of UBC13 but not the other proteins shifted in an OspI dose-dependent manner. Furthermore, OspI could interact with His-UBC13, suggesting that OspI targeted UBC13 to alter its negative surface charge. Hence, we used LC-MS/MS to examine how OspI post-transcriptionally modifies UBC13. We determined that two overlapping tryptic-digested peptides (WSPALQIR and DKWSPALQIR) of UBC13 were deamidated at glutamine (Q, Gln) 100 to glutamic acid (E, Glu) 100 by OspI. To confirm that OspI deamidates UBC13, we created UBC13 (Q100E), which underwent the same mobility shift as UBC13 that was modified at Gln 100 by Ospl, but not OspI C62A, H145A or D160A. Consistently, endogenous UBC13 was deamidated in HeLa cells at 10 min after infection with YSH6000 and ΔospI/ospI but not ΔospI or ΔospI/ospI (C62S). Of note the co-crystal structures of UBC13 and the Zn-finger of TRAF6 indicated that Gln100 of UBC13 was proximal (13 amino acids behind) to the catalytic pocket, but also located near the interface between UBC13 and the TRAF6 Zn-finger. Thus, we further characterized the Ub-conjugating E2 activity of UBC13 (Q100E) using an in vitro ubiquitination assay. The efficiency of ubiquitin chain formation catalyzed by UBC13 (Q100E) with TRAF6 was greatly decreased compared with wild-type UBC13. In an NFκB reporter assay, UBC13 (Q100E) acted as a dominant-negative, in which UBC13 (Q100E) suppressed the NFκB activity that was stimulated by PMA, TRAF6 and infection, but not TNFα, in a dose-dependent manner. These results confirmed that OspI has deamidase activity against UBC13 Gln100, which allows OspI to dampen the TRAF6-NFκB pathway.
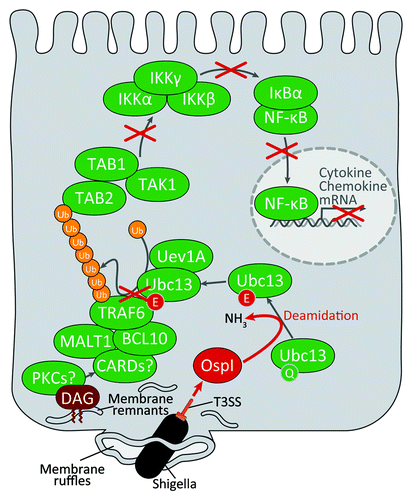
In summary, we identified OspI as a new T3SS effector that specifically targets TRAF6-dependent acute inflammatory signaling during S. flexneri invasion of epithelial cells. OspI selectively deamidates UBC13 to inactivate the E2 ubiquitin ligase activity that is required for TRAF6 polyubiquitination, allowing S. flexneri to block acute NFκB-mediated inflammatory responses at the early stage of epithelial invasion (). Recent studies reported that the cycle inhibiting factor (Cif) from enteropathogenic E. coli and the Cif homolog from Burkholderia pseudomallei (CHBP) selectively deamidate ubiquitin-like protein NEDD8 and ubiquitin, which abolishes Cullin-RING ubiquitin ligase activity. The selective Cif-mediated deamidation of NEDD8 is linked to the ability of some enteropathogenic E. coli strains to induce cell cycle arrest and actin stress fiber formation. In our study, we performed LC-MS/MS analysis and showed that OspI and Cif do not have deamidase activity against ubiquitin and UBC13, respectively. We also performed a native PAGE analysis to further confirm the substrate specificity and dose-dependency of the deamidation activity of OspI and Cif. We found that Cif deamidates UBC13 and ubiquitin, but only at high concentrations. Although Cif preferentially deamidates NEDD8, Cif also deamidates ubiquitin, but only at high concentrations. Therefore, together these studies suggest that OspI and Cif have distinct substrate specificities. Our structural and functional analyses of OspI strongly indicate that OspI is a unique T3SS effector that dampens TRAF6-dependent inflammatory signaling in response to bacterial internalization in epithelial cells.
Figure 2. Shigella inhibits acute inflammatory responses at the initial stage of infection. Shigella invades the host cell by macropinocytosis and quickly escapes from the phagosome into the cytoplasm. The phagosome membrane fragments are produced by Shigella upon escape in the host cytoplasm. DAG accumulates around the bacterial entry site. This accumulation activates the diacylglycerol-CBM complex-TRAF6-NFκB signaling pathway. Shigella OspI is delivered via the type III secretion system during bacterial invasion. OspI acts as a glutamine deamidase and selectively deamidates Gln100 to Glu100 in UBc13, severely impairing the E2 ubiquitin conjugating activity of Ubc13, which is required for the activation of the TRAF6-NFκB pathway.