Both pathogenic and non-pathogenic microbes express common microbe-associated molecular patterns (MAMPs) that can activate innate pro-inflammatory responses. There is evidence to suggest that host immune systems are adapted to minimize unwarranted, potentially damaging pro-inflammatory responses, e.g., to commensals at muscosal surfaces where very high microbe loads are found, while maintaining the ability to mount aggressive responses to invading pathogens. The recognition of bacterial secretion systems by host innate immune pathways may represent one mechanism used by host immune cells to discriminate between pathogen and non-pathogen, or strains with reduced virulence potential. Most recently, it has been shown that the type IV secretion system (T4SS) encoded by virulent cagPAI-positive strains of the pro-carcinogenic pathogen Helicobacter pylori induces the expression of microRNA-155, a known mediator of innate immunity that is also implicated in oncogenesis. The T4SS-specific activation of miR-155 occurred independently of TLR and NOD1/2 pattern-recognition receptor (PRR) signaling. We discuss the potential role of the T4SS-dependent activation of miR-155 as a pathogen-specific immune response and the possible implications of this in the context of gastrointestinal macrophage inflammatory anergy, a phenomenon in which PRR signaling is inhibited in gastrointestinal resident phagocytes. We also touch on the observed anti-apoptotic role of miR-155 during H. pylori infection, and speculate as to its possible pathological consequences.
Helicobacter pylori is an ancient (Moodley et al., PLoS Pathog 2012) Gram-negative bacterial pathogen that colonizes the gastric mucosa of humans, its only natural host. It is a ubiquitous pathogen that infects some 50% of the global population. Primary infection normally occurs in childhood and persists for the lifetime of the host. Infections are characterized by gastritis (frequently asymptomatic) in all infected individuals, which can progress to peptic ulcer disease (10–15% of infected individuals) or, through a series of histologically defined stages, to non-cardia gastric adenocarcinoma (1–3% of infected individuals) (Fox and Wang, J Clin Invest 2007; Parsonnet et al., New Engl J Med 1991). Infection is also associated with B cell mucosa associated lymphoid tissue (MALT) lymphoma (0.2% of infected individuals) (Capelle, Eur J Can 2008). Thus far, there is no vaccine that can induce protective immunity and although drug regimes that include antibiotics can clear infection and peptic ulcer disease and regress MALT lymphoma, individuals that had evidence of histological progression to intestinal metaplasia prior to infection clearance probably remain at increased risk of gastric cancer (Zullo et al., W J Gastroenterol Oncol 2012). An immune response is mounted by the host to infection, but this is almost always inadequate in terms of infection clearance, and the chronic inflammatory response that can result is widely accepted to induce immunopathology and underlie oncogenesis. The pathological outcome of H. pylori infection is likely dependent on a number of bacterial, host and environmental determinants, and the complex interplay between these factors (Pritchard and Crabtree, Curr Opin Gastroenterol 2006). There is much still to be clarified as our current understanding of H. pylori virulence and the role of deregulated inflammatory responses during pathogenesis is incomplete.
H. pylori virulence is associated with an ~40-kb horizontally acquired genomic pathogenicity island of unknown origin, the cagPAI, which encodes a type four secretion system (T4SS) and the prototypical H. pylori secreted virulence protein, CagA, among others (Amieva et al., Science 2003). In epithelial cell lines, experimental animal infection models and clinically, infection with cagPAI-positive strains is associated with increased inflammatory responses and risk of gastric cancer (Crabtree et al., J Clin Pathol 1995; Parsonnet et al., Gut 1997; Rieder et al., Gastroenterol 2005).
The host immune response to H. pylori infection is also strongly implicated in pathogenesis of infection (Roth et al., J Immunol 1999). An unresolved inflammatory response most likely serves to increase infiltration of monocytes and neutrophils from the circulation, creating an environment that primes T cells for a Th1/Th17 dominant T cell adaptive immune response that appears to be responsible for attempts to control and clear infection, and at the same time, for immunopathology and associated disease (Sayi et al., J Immunol 2009). Prominent early innate defense mechanisms are directed against H. pylori by gastric epithelial cells and tissue resident innate immune cells such as macrophages and dendritic cells (DCs). Detection of microbe associated molecular patterns (MAMPs) by pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), nucleotide oligomerization domain (NOD) receptors and the retinoic acid inducible gene 1 receptor (RIG-I) is an important component of the host innate immune response to H. pylori in the gastric mucosa and results in the induction of pro-inflammatory cytokines, particularly in macrophages and DCs. Isolated DCs have been shown to upregulate inflammatory cytokines such as IL-6 in response to H. pylori infection in a MyD88-dependent manner. H. pylori MAMPs activate extracellular TLR-2 and -4 (LPS), and endosomal TLR-9 (bacterial DNA) to induce the nuclear factor kappa B (NFκB) pro-inflammatory pathway (Rad et al., Gastroenterol 2009; Obonyo et al., Infect Immun 2007) (). H. pylori RNA can also activate the soluble cytosolic RIG-I receptor in DCs to induce IFN-γ responsive genes (Rad et al., Gastroenterol 2009) and bacterial peptidoglycan activates the cytosolic NOD1 receptor to induce IL-8 production in gastric epithelial cells in a cagPAI-dependent manner, which contributes to inflammatory responses (Viala et al., Nat Immunol 2004). Also implicated in the innate immune response to H. pylori is infection-induced cellular microRNA (miRNA) expression (Xiao et al., J Infec Dis 2009). MicroRNAs are short non-coding RNAs that are post-transcriptional regulators of gene expression. Typically, they bind to the 3′-UTRs of their mRNA targets to inhibit gene expression. The modulation of cellular miRNA expression in response to bacterial infection is, potentially, a common theme in host-pathogen systems that has broad implications for immunity and disease. Through their regulation of key inflammatory signaling pathways—in particular NFκB—and cytokine gene expression, miRNAs are increasingly being implicated as important mediators of host immune responses to bacterial and viral pathogens (Gantier, J Interferon Cytokine Res 2010; Eulalio et al., RNA Biol 2012), and as potential disease loci in humans. MicroRNA expression can be activated by PRRs; for example, LPS-mediated stimulation of TLR4 has been shown to induce the expression of miR-132, miR-146a/b and miR-155. The NFκB pathway is also implicated in the expression of miRNAs, including miR-155 (Eulalio et al., RNA Biology 2012). miR-155 may be of particular relevance to infection and immunity as it is induced in macrophages in response to TLR ligands and Gram-negative and Gram-positive pathogens and is widely implicated in inflammatory responses and innate immunity; for example, it was found to be induced in a TLR-dependent manner during infection of macrophages by the Gram-negative intestinal pathogen Salmonella enterica and was crucial for normal immune function during experimental Salmonella infection of mice, and for the induction of protective immunity (Rodriguez et al., Science 2007). miR-155 was also among the most significantly upregulated miRNAs during infection with the Gram-positive pathogen Listeria monocytogenes in macrophages (Schnitger, PLoS One 2011). The induction of miRNAs, including miR-155, has also been reported during L. monocytogenes infection of epithelial cells (Izar et al., Int J Mol Sci 2012). Interestingly, the regulation of miRNA expression in that study was dependent on L. monocytogenes virulence factors and the sub-cellular localization of bacteria, which suggests that, at least for intracellular pathogens, this may provide an additional level of host-pathogen cross talk. miR-155 is upregulated in a TLR-dependent manner during H. pylori infection in a number of different settings (Fassi Fehri et al., PLoS One 2010). Interestingly, miR-155 is associated with both pro-inflammatory responses and oncogenesis, warranting further investigation of the role of this miRNA in H. pylori infection-induced pathology.
Figure 1. The proposed T4SS-dependent activation of miR-155 expression in macrophages during H. pylori infection. The T4SS, specific to virulent H. pylori strains, induced NFκB dependent miR-155 expression, in addition to the known activation by H. pylori MAMPs via TLR and NOD1/-2 pattern recognition receptor signaling. T4SS-dependent miR-155 expression, represented by (?), may be directly dependent on an unidentified cellular receptor, or on an indirect activation pathway. miR-155 was found to target a number of pro-apoptotic genes in macrophages, and conferred an anti-apoptotic effect upon infection in the presence of a DNA damage inducing reagent. It is speculated that miR-155 expression could prolong macrophage survival in the inflammatory microenvironment, which could contribute ultimately to the deregulation of pro-inflammatory responses, increased expression of proinflammatory mediators e.g., IL-8 and IL-6 and pathology progression observed during H. pylori infection. MAMP, microbe-associated molecular pattern; NOD, nucleotide oligomerization domain; ROS, reactive oxygen species; RNS, reactive nitrogen species; T4SS, type IV secretion system; TLR, Toll-like receptor.
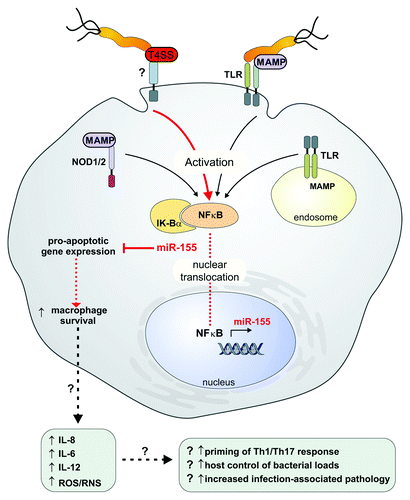
Recently, we observed that the H. pylori T4SS pathogenicity determinant upregulated miR-155 expression during experimental infection of primary macrophages deficient in TLR and NOD1/2 signaling (Koch et al., Proc Natl Acad Sci U S A 2012). This latest finding that H. pylori modulates the expression of miR-155 by a mechanism partly dependent on the T4SS, and independent of TLR/NOD1/-2 receptor recognition is intriguing. Since only the more virulent cagPAI positive H. pylori strains contain the T4SS, this could suggest that the T4SS acts as a MAMP that provides a critical function for the host innate immune system: the ability to distinguish between pathogenic strains, and less pathogenic strains and commensals. Both pathogenic and non-pathogenic bacterial populations possess common PRR-activating MAMPs such as lipid A, and so a pivotal question in biology is whether immune systems can discriminate between pathogens and commensals, and if they do, then how? There is mounting evidence to suggest that host cells are able to distinguish between pathogenic and less pathogenic strains and commensals. One possible mechanism may involve exploiting the PRR-independent detection of pathogen-specific features, including secretion systems, to ensure that inflammatory responses are commensurate with threat level, since in addition to common MAMPs, pathogenic species typically also express virulence factors and machinery, specific combinations of which may be unique to pathogenic strains (Blander and Sander, Nat Immunol 2012). It has been postulated that pathogens display particular signatures, or “patterns of pathogenesis,” that can act as PRR-independent activators of immune responses, which may then synergize with PRR-mediated signaling to optimize and target the response to pathogen infection (Fontana et al., PLoS Path 2011; Vance et al., Cell Host Mic 2009). Examples of pathogen-specific patterns of pathogenesis typically involve initiation of general perturbations to cellular function/breaching of cell barriers, and may include deregulation of the actin cytoskeleton, induction of cell death (Vance et al., Cell Host Mic 2009) and inhibition of host translation (Fontana et al., PLoS Path 2011). It has been noted that patterns of pathogenesis may distinguish viable from non-viable bacteria, and the term vita-PAMP was coined to describe bacterial MAMPs that stimulate a host immune response only when pathogens are viable (Sander et al., Nature 2011; Vance et al., Cell Host Mic 2009). The ability to distinguish dead from viable bacteria could be important for scaling of immune responses in accordance with perceived threat level. Bacterial virulence factors such as secretion systems, and secreted effector proteins, have themselves been experimentally implicated as having differing functional roles in the PRR-independent activation of host immune responses, and may act as vita-PAMPs in this capacity. Evidence suggests that secretion systems can directly activate PRR-independent immune responses, i.e., via an interaction between the secretion system and a cellular factor/receptor, or indirectly, which may not involve a cellular receptor. Secretion system, specifically type III secretion system (T3SS) and T4SS, dependent activation of the NLRC4 inflammasome may be important for distinguishing pathogen from non-pathogen, for example; this has been reported to occur indirectly, e.g., via the secretion-system mediated delivery of flagellin into host cells, and directly, e.g., via the interaction of the relatively highly conserved rod region of the T3SS from a number of pathogenic species including Shigella flexneri, S. Typhimurium and Burkholderia pseudomallei (Miao et al., Proc Natl Acad Sci U S A 2010). Recently, it was demonstrated that in murine intestinal phagocyte populations, pathogenic Salmonella and Pseudomonas bacteria induced the expression of the pro-inflammatory cytokine IL-1β through activation of the NLRC4 inflammasome, whereas commensal bacteria did not. The production of IL-1β was dependent on a functional T4SS or T3SS and was required for infection clearance (Franchi et al., Nat Immunol 2012); the authors did not describe whether the secretion systems played a direct or indirect role in inflammasome activation. The molecular mechanism by which the T4SS activated NFκB-dependent miR-155 expression in a TLR- and NOD1/2-independent manner in macrophages during H. pylori infection remains to be resolved, although the secreted H. pylori virulence factor CagA was excluded as having a role—of course this does not rule out the possibility that other secreted factors are involved. Whether the T4SS mediated induction of miR-155 observed in the latest study requires the direct interaction of the T4SS with an as yet unidentified host cell receptor, or whether there is an indirect mechanism responsible for NFκB activation and miR-155 induction, remains to be determined. It is conceivable that there is a direct interaction between the T4SS and a host cell receptor in macrophages. Certainly, the integrin α5β1 has been suggested elsewhere to be a host cell receptor for the H. pylori T4SS (Kwok et al., Nature 2007) and virulence factors from other pathogens are also known to exploit integrin α5β1 as a receptor; for example, the surface protein Td92 of the periodontopathogen Treponema denticola binds to α5β1 and activates the NLRP3 inflammasome and pro-IL-1β transcription via an NFκB dependent pathway (Jun et al., Immunity 2012).
The contribution of the T4SS to miR-155 expression was significant but modest in macrophages in vitro. We speculate, however, that this effect may take on new relevance in tissue resident macrophage populations in the gastric muscosa, which are phenotypically distinct from blood monocytes. Gastrointestinal tissue-resident macrophages—the largest population of macrophages in the body—reportedly exhibit inflammatory “anergy” under conditions of tissue homeostasis. Anergic macrophages are typically downregulated for PRR signaling, including TLR, but retain phagocytic properties (Smythies et al., J Clin Inv 2005). Inflammatory anergy is assumed to be an immunotolerance mechanism that regulates immune responses at mucosal surfaces, which present particular challenges as they are bombarded with foreign antigens from various sources. We suggest that the ability of macrophages to detect the H. pylori T4SS pathogenicity determinant, independent of TLR signaling, may contribute to the pro-inflammatory response to H. pylori in the context of gastric mucosal macrophage inflammatory anergy. Interestingly, Franchi et al. observed the bacterial secretion-system dependent induction of the inflammasome in anergic murine intestinal phagocyte populations (Franchi et al., Nat Immunol 2012). However, whether the H. pylori T4SS specific induction of miR-155 occurs in anergic macrophages or indeed, whether it contributes to inflammatory responses during H. pylori infection requires further investigation.
A number of cellular microRNAs are upregulated, and downregulated, in the gastric mucosa in response to H. pylori infection (Matsushima et al., Int J Cancer 2011; Fassi Fehri et al., PLoS One 2010). Concerning H. pylori-induced pathology, miR-155 is of particular interest as it is widely implicated in inflammatory responses and cancer and, in particular, is observed to be overexpressed in gastric cancer and B cell lymphomas. Moreover, during in vitro and in vivo investigations, miR-155 was consistently upregulated in response to H. pylori infection in a number of non-hematopoietic and hematopoietic cells. In gastric epithelial cell lines and gastric mucosal tissues, miR-155 was upregulated by the NFκB and AP-1 pathways in response to infection (Xiao et al., J Infect Dis 2009); miR-155 overexpression in this system reportedly inhibited the expression of IL-8. We have shown that miR-155 is upregulated in response to H. pylori infection of a human T-cell line and murine macrophages, and in gastric mucosal biopsies from human volunteers (Fassi Fehri et al., PLoS One 2010). The role of miR-155 may best be described as “immunomodulatory” during infection since NFκB pathway genes are also miR-155 targets; thus, the regulation of this miR-155 is under negative feedback control, which may ultimately serve to “finetune” inflammatory responses. One can only speculate as to the role of miR-155 in macrophages in vivo during infection, particularly as the role of macrophages in the host response to H. pylori infection and infection-induced pathology is in general unclear. In the latest study, comparative microarray analysis of miR-155−/− and wild type primary murine bone marrow derived macrophages showed that miR-155 regulated the expression of a large subset of genes that are involved in cell-death. Indeed, when macrophages were stressed with the DNA-damaging reagent cisplatin during H. pylori infection, miR-155 protected the cells against apoptosis, and the overall effect of miR-155 in this system was anti-apoptotic. We suggested that this could be potentially biologically significant in vivo for prolonging macrophage survival and, thus, proinflammatory potential in the inflammatory microenvironment. This is anecdotally supported by the observation that mice chemically depleted of macrophages exhibit reduced pathology during H. pylori infection, despite having comparable bacterial loads to wild-type mice (Kaparakis et al., Infect Immun 2008). The role of miR-155 expression in other cell types during H. pylori infection in vivo is also ambiguous; to date, results in animal models have shown that miR-155 expression in T cells controls bacterial load. Interestingly, in this model miR-155−/− mice were significantly protected against H. pylori induced pathology and showed fewer precancerous lesions despite high bacterial loads compared with wildtype mice. Reduced ability to control infection and the decreased pathology in miR-155−/− mice were both largely attributed to defective production of IL-17 and IFN-γ. Together, these data suggest that miR-155 induced during H. pylori infection has a key role in both infection control and pathogenesis (Oertli et al., J Immunol 2011); however, the current experimental evidence highlights the need for in vivo clarification of the functional consequences of infection-induced changes in miR-155 expression profiles in the different cell types present in the gastric mucosa.
It is clear that microbes can manipulate cellular miRNA profiles to profoundly alter host biology; moreover, altered or deregulated miRNA profiles in host cells during infection may prove to be a common pathogenesis mechanism, with implications for clinical management of infection-induced pathologies. The latest finding that the T4SS specifically upregulates miR-155 in macrophages during infection with H. pylori suggests that, similar to other innate immune responses that are pathogen specific, the T4SS provides macrophages with an additional level of discrimination for mounting a response to cagPAI-positive H. pylori strains. Mechanistically, how the T4SS activates the cellular miR-155 response, and whether this response in macrophages contributes to deregulated inflammatory responses during infection and disease phenotypes, will be interesting topics for future research into the virulence potential of H. pylori.
Acknowledgments
Work in the Meyer laboratory for the original publication was funded by the Sixth Research Framework Programme of the European Union, Project SIROCCO (LSHG-CT-2006-037900). M.K. was funded by the International Max Planck Research School for Infectious Diseases and Immunology. The authors would like to thank Diane Schad for her expert assistance with preparation of the figure.