Abstract
Molecular mimicry of host proteins is a common strategy adopted by bacterial pathogens to interfere with and exploit host processes. Despite the availability of pathogen genomes, few studies have attempted to predict virulence-associated mimicry relationships directly from genomic sequences. Here, we analyzed the proteomes of 62 pathogenic and 66 non-pathogenic bacterial species, and screened for the top pathogen-specific or pathogen-enriched sequence similarities to human proteins. The screen identified approximately 100 potential mimicry relationships including well-characterized examples among the top-scoring hits (e.g., RalF, internalin, yopH, and others), with about 1/3 of predicted relationships supported by existing literature. Examination of homology to virulence factors, statistically enriched functions, and comparison with literature indicated that the detected mimics target key host structures (e.g., extracellular matrix, ECM) and pathways (e.g., cell adhesion, lipid metabolism, and immune signaling). The top-scoring and most widespread mimicry pattern detected among pathogens consisted of elevated sequence similarities to ECM proteins including collagens and leucine-rich repeat proteins. Unexpectedly, analysis of the pathogen counterparts of these proteins revealed that they have evolved independently in different species of bacterial pathogens from separate repeat amplifications. Thus, our analysis provides evidence for two classes of mimics: complex proteins such as enzymes that have been acquired by eukaryote-to-pathogen horizontal transfer, and simpler repeat proteins that have independently evolved to mimic the host ECM. Ultimately, computational detection of pathogen-specific and pathogen-enriched similarities to host proteins provides insights into potentially novel mimicry-mediated virulence mechanisms of pathogenic bacteria.
Introduction
Molecular mimicry can be broadly defined as sequence or structural resemblance between microbial and host molecules. This has been studied extensively within the context of autoimmunity, whereby similarities between foreign and self molecules can lead to cross-reactive epitopes and ultimately autoimmune disease.Citation1-Citation5 However, there is a growing body of evidence that molecular mimicry of host proteins is a broader strategy adopted by bacterial pathogens to exploit and subvert host processes during infection and plays a role in a wide range of virulence pathways, including pathogen recognition and binding to human cells, evasion of the host immune response, and intracellular survival in host immune cells.Citation6-Citation10
Mimics within pathogens are thought to originate through two evolutionary mechanisms. Pathogen genomes can obtain host genes directly through lateral transfer (reviewed in Koonin et al.Citation11). Such cases frequently have detectable homology between pathogen and host proteins, a complex sequence or domain composition, and limited occurrence of the mimic in one or a small number of pathogenic species. For example, Coxiella burnetii, the causative agent of human Q fever, encodes two eukaryote-like sterol reductases. These mimics may play a role in formation of the cholesterol-rich Coxiella parasitophorous vacuole,Citation12 which serves as a barrier to sequester nutrients and ions and also facilitates pathogen survival inside the host cell. These enzymes are extremely rare in prokaryotes and are thought to have arisen in Coxiella by lateral transfer from a eukaryotic source.Citation13
A second possible mechanism is convergent or parallel evolution of a pathogenic protein toward resemblance of a host protein.Citation7,Citation10,Citation14 Here, over time, co-evolutionary forces generate pathogen proteins that resemble host proteins structurally, or resemble smaller sequence fragments of host proteins, without homology between the pathogen and host proteins.Citation7 For example, enterohemorrhagic Escherichia coli (EHEC) secretes a type III effector (EspFU) into human host cells, which stimulates actin polymerization by interacting with host WASP proteins.Citation15 Exploitation of host functions is achieved through subtle structural mimicry of the host WASP autoinhibitory helix, but there is no detectable sequence similarity between the two proteins. By stimulating actin polymerization, EspFU mediates attachment of EHEC to host epithelial cells, which is critical to its virulence mechanism. Another example of convergent evolution is that of the Yersinia effector protein, invasin, which has evolved to mimic the integrin-binding surface of fibronectin.Citation16 This surface mediates high affinity binding to β1 integrins on host M cells, which induces cytoskeletal reorganization and allows the pathogen to gain entry into the host cell.
Though there are exceptions (e.g., see Graham et al.Citation17), pathogen mimics tend to function similarly to their human counterparts. For instance, through mimicry of human guanine-exchange factors (GEFs), the Legionella effector RalF functions as a GEF in the host and recruits ADP-ribosylation factor (Arf) to manipulate host vesicular trafficking.Citation18 Through mimicry of human tyrosine phosphatases, Yersinia YopH dephosphorylates a number of human proteins including p130Cas that leads to inhibition of phagocytosis (reviewed in Stebbins and GalanCitation7 and Knodler et al.Citation8). Furthermore, internalin virulence factors are composed of leucine-rich repeats (LRRs) with binding surfaces like eukaryotic LRRs, and play a role in adherence and invasion of host cells.Citation7,Citation19,Citation20
To date, discovery of pathogen mimics has been done largely on a case-by-case basis, and it is possible that there exist many additional mimics that may be detectable through computational methods. In previous work, for instance, we identified sequence and structural similarities between clostridial toxins and mammalian collagens, from which we hypothesized that collagen may be an additional mimicry target of pathogenic bacteria,Citation21 which could play a role in adhesion of pathogens to the host extracellular matrix. However, detection of sequence similarity between host and pathogenic proteins is by itself not indicative of mimicry or pathogen-specific exploitation of host functions.
Here, motivated by our previous work and the broad goal of detecting host-pathogen mimicry at a genomic scale, we performed an analysis of bacterial pathogen vs. non-pathogen proteomes and compared their similarities to the host (i.e., human) proteome. We screened for cases where the detected similarities to host proteins are pathogen-specific or are enriched in a variety of pathogenic species compared with non-pathogens, thus producing a list of candidate pathogen mimics and their human targets. It is important to note that while the analysis is based on the human proteome, host specificity of the predicted mimics toward human is not certain, and the predictions may reflect mimicry of proteins from alternative eukaryotic hosts. A similar approach has been used to screen for molecular mimicry candidates in protozoan parasites,Citation22 yet to our knowledge a large-scale computational analysis has not been performed for human pathogenic bacteria.
Ultimately, our results provide additional evidence that collagens and extracellular matrix proteins in general are targets of mimicry by a range of pathogenic bacteria. Moreover, we report the unexpected result that such mimics have evolved independently in a range of bacterial pathogens through separate amplifications of short peptide repeats. In addition to extracellular matrix proteins, the screen predicted numerous known and potentially novel mimicry relationships that are candidates for future experimental investigation.
Results
Detecting pathogen mimics of human proteins by comparative proteome analysis
We developed and applied a computational pipeline (outlined in ) to identify potential pathogen mimics of human (host) proteins using comparative proteome analysis. First, proteome sequence data was retrieved for human and 128 bacterial species including 62 bacterial pathogens of humans and 66 non-pathogens as annotated by the Comprehensive Microbial Resource (CMR).Citation23 An all-by-all BLASTCitation24 analysis was performed, in which all human proteins were searched against all bacterial proteomes, and hits with E-values < 1e−06 were collected and compared between the two groups ().
Figure 1. Computational pipeline for detection of molecular mimicry candidates in human pathogenic bacteria.
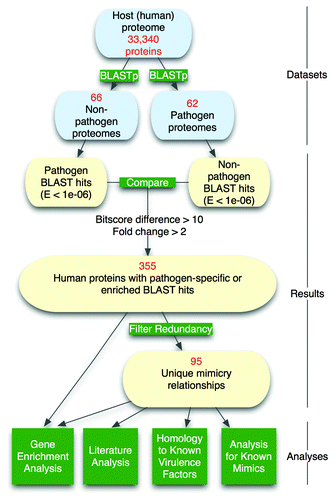
From the set of 33 340 human proteins, 9149 (27.4%) had BLAST matches with E < 1e−06 in one or more bacterial proteomes (Fig. S1A). For the average human protein, the frequency of pathogen vs. non-pathogen genomes containing a match, and the top pathogen vs. non-pathogen alignment score (bitscore) and E-value, was highly similar (Fig. S1B–E). Moreover, a larger fraction of the human proteome was similar to proteins in the non-pathogen set (Fig. S1A), which may reflect pathogen-associated genome reduction.Citation25
While molecular mimics may be encoded by pathogenic and non-pathogenic species, and serve different biological purposes (e.g., virulence, mimicry of immune epitopes, and survival of commensal bacteria inside host), we aimed to identify mimics that may play specific roles in pathogen virulence. Here, we use the definition of pathogen mimics as bacterial pathogen-encoded proteins that share significant similarities with host proteins for the purposes of interacting or interfering with host machinery for the pathogen’s benefit.Citation10,Citation22 To identify such a subset, we further processed this list of bacteria–human protein similarities to identify those specific to or enriched in pathogens and diminished or completely absent in the non-pathogen group (controls), and thus indicative pathogen–host specificity and potential molecular mimicry. This involved applying the following criteria: that a hit is specific to or at least 2-fold enriched in pathogen species, and that a top pathogen hit has a greater alignment score than the corresponding top non-pathogen hit (bitscore difference >10, see Methods) (). These parameters capture a subset of potential mimics within the distributions shown in Figure S1C–E.
Applying these filters resulted in a final list of 355 human proteins predicted as potential targets of pathogen molecular mimicry (Table S1A). These predicted mimicry relationships occurred in a small number of bacterial species (average = 3.2 species, 2.5%) compared with that observed for non-mimics (average = 36.8 species, 28.8%), which as expected appears to contain all ubiquitous proteins in the data set (2333 found in human and a majority of bacterial species, 208 conserved across all species).
The 355 human proteins were predicted targets of 231 total pathogen proteins from 53 pathogen species (Table S1B). We also removed redundancy within this set (see Methods) in order to generate a smaller list of 95 highly unique relationships (Table S1C) for subsequent analysis (see Methods). The top 25 most unique mimicry predictions are listed in . As explored in later sections, the top predictions include extracellular matrix proteins (collagen and leucine-rich repeat proteins), as well as several virulence factors and known examples of molecular mimicry ().
Table 1. Top 25 unique predictions of molecular mimicry relationships.
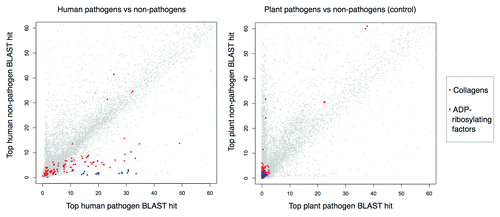
displays a plot of the top BLAST matches to each human protein in pathogen vs. non-pathogen proteomes. Similarities to the well-known mimicry targets, guanine-exchange factors,Citation26 are shown in blue and are clear outliers in this distribution, which serves as a positive control. Similarities to human collagens, which correspond to the top predicted mimicry relationships (), are also indicated in red, and similarly display elevated scores in the pathogen set. As an additional control, we performed the same computation using plant pathogen/non-pathogen definitions (6/16 respectively), and this signal was absent (, right).
Figure 2. Top BLAST matches for human proteins in pathogen vs. non-pathogen proteomes. Left: −log10E-values for top BLAST matches to human proteins in 62 human pathogens vs. 66 non-pathogens. Right: −log10E-values for top BLAST matches to human proteins with different host/pathogen definitions (6 plant pathogens vs. 16 non-pathogens). Values above ~60 are not shown. Collagens (top detected mimicry relationship) and ADP-ribosylating factors (positive control mimicry relationship) have pathogen-elevated E-value distributions.
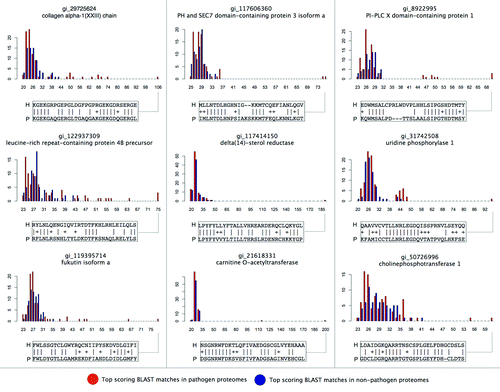
The pathogen vs. non-pathogen bitscore distributions for nine of the top-scoring hits from are shown in . In each case, the overall bitscore distributions for both pathogen and non-pathogen proteomes are similar to the extreme value distributions that might be expected by random alignments, but also contain pathogen proteins with considerably elevated similarities to a human protein, indicative of mimicry. Some of the detected mimics occur in a few pathogens (e.g., delta sterol reductase homolog exclusive to Coxiella burnetii), while others occur more broadly throughout a range of pathogen species (e.g., putative mimics of collagens and leucine-rich repeat proteins, ).
Figure 3. Top pathogen vs. non-pathogen protein similarities to a selected set of predicted human mimicry targets. Predicted human mimicry targets were selected from the top 25 detected relationships (), and the top BLAST matches by bitscore (x-axis) in pathogen vs. non-pathogen proteomes (frequency on y-axis) have been plotted. In each case, it can be seen that a subset of pathogen proteomes encode putative mimics that exhibit much greater similarities to human proteins than similarities found in non-pathogen proteins. A selected portion of the alignment is shown for the top-scoring pathogen mimic in each case. See Data File S1 for additional details regarding pairwise alignments.
Top predictions include known mimics and are enriched in virulence-related functions
We then examined the list of predicted mimicry relationships in terms of known mimics, homology to virulence factors, statistically enriched functions, and comparison with literature. These analyses suggest that the predictions are enriched in mimicry-mediated virulence mechanisms of bacterial pathogens, and include both known and putative novel mimicry relationships.
Known mimics
Prediction #3 corresponds to detected similarity between a human leucine-rich repeat protein (gi 122937309) and a leucine-rich repeat protein from Streptococcus pyogenes (SpyM3_1561) () as well as Listeria internalin virulence factors (.g., lmo0801 and LMOh7858_0295 [Table S1A]). This reflects the established example of human LRR mimicry by internalin.Citation7,Citation19,Citation20 Predictions #17 and #18 are detected relationships between two human guanine exchange factor (GEF) containing proteins and the pathogen proteins RalF (Legionella pneumophila) and sec7 (Rickettsia prowazekii), and thus reflect GEF mimicry.Citation18 Prediction #31 corresponds to detected similarity between a human tyrosine phosphatase (gi 108802617) and yopH from Yersinia pseudotuberculosis, another known case of pathogen-specific molecular mimicry.Citation7,Citation8
Homology to virulence factors
Sixty-one of 95 predicted mimics (non-redundant set) were detected as homologous (BLAST E-value < 1e−06) to known virulence factors from the MvirDB,Citation27 suggesting possible roles in mimicry-mediated virulence. These include the Legionella LepB effector (prediction #7, similar to numerous human coiled coil proteins) indicating possible functions in regulation of secretory traffic, Listeria PlcA (prediction #19, similar to human PI-PLC), and Helicobacter and human fucosyltransferase (prediction #68, FucT) (; Table S1). Helicobacter FucT is another known case of mimicry as its produces the lipopolysaccharide component Lewis X trisaccharide, which is thought to mimic host sugars to escape immune detection.Citation28
Enriched virulence-related functions
Gene enrichment analysis was then performed using DAVIDCitation29 to identify statistically enriched functions and protein families among the full set of predicted human mimicry targets (; Table S2A). The top four enriched function terms were: “Extracellular matrix” (Benjamini P = 3.39E−42), collagen (P = 3.11E−29), “Extracellular matrix structural protein” (P = 2.56E−24), and “ARF guanyl-nucleotide exchange factor activity” (P = 2.11E−19) (Table S2A). Other intriguing top enriched terms included “O-acyltransferase activity” (P = 2.08E−06), “cell adhesion (P = 2.95E−06), and “inflammation mediated by chemokine and cytokine signaling pathway” (P = 6.84E−03). Terms that were highly ranked and functionally relevant but of weaker statistical significance included “Iκ kinase/NFκB cascade” (P = 1.18E−02), “lysosome” (P = 1.38E−02), “Toll-like receptor pathway” (P = 5.15E−02), and interaction with host (P = 9.30E−02). Thus, the top enriched terms appear to be consistent with virulence related mechanisms of pathogenic bacteria. As a comparison, we also applied the same analysis to phytopathogens (578 mimics detected, data not shown), which identified different enrichment categories. The top two functional enrichments for predicted mimics from phytopathogens were “apoptosis” (P = 5.54E−27) and “programmed cell death” (P = 1.42E−25). Indeed, induction of apoptosis is a known virulence mechanism of plant pathogens,Citation30 suggesting that the approach may be applicable to different host-pathogen relationships.
Table 2. Function enrichment analysis of predicted mimicry candidates
Comparison with literature
We then analyzed the top 95 unique detected mimicry relationships (non-redundant set) for additional literature supporting potential roles in virulence. Thirty-three of these relationships (~35%) were found to be highly suggestive of mimicry-mediated virulence mechanisms with support from existing literature (). Known mechanisms of pathogen exploitation of host cells that are also reflected throughout the detected relationships include pathogen-specific modulation of host lipid metabolism, modulation of the host nucleotide pool and induction of apoptosis, and cell adhesion. The remaining 65 cases may represent previously uncharacterized mimicry relationships, and provide numerous targets for investigation of pathogenicity mechanisms.
Table 3. Predicted mimicry candidates in human pathogenic bacteria and potential roles in virulence
Numerous detected mimicry relationships and function enrichments (Table S2) are linked to lipid metabolism. Putative mimics affecting host lipid metabolism include detected pathogen counterparts of human cholinephosphotransferase, PCTP like protein, fukutin, acyltransferases, phospholipase A2, carnitine O-palmitoyltransferase, and sterol reductase (). For example, Mycobacterium pneumonia is known to incorporate host lipids (e.g., phosphatidylcholine),Citation68 and has been shown to modulate host sphingolipid metabolism.Citation45 A human-like carnitine O-palmitoyltransferase (CPT) was detected in Mycobacterium pneumonia, which may play a role in these functions.Citation44 As another example, part of the virulence mechanism of Bacillus anthracis involves escape from host macrophages, but the mechanism is largely unknown.Citation69 A human-like phospholipase A2 was detected in Bacillus anthracis (BA_3805), which, in other pathogens, has been shown to play a role in phagosome escape as well as entry and lysis of host cells.Citation64,Citation65
Cells damaged by invading pathogens release nucleotides into the external environment, which act as “danger signals” and stimulate pathogen-killing immune responses,Citation37,Citation70 or induce apoptosis.Citation71 Detected mimicry relationships indicative of host nucleotide pool modulation include the previously studied Legionella lpl1869 with detected similarities to the human NTPDase CD39,Citation60 and proteins similar to human P-loop NTPases and ATPases from Vibrio and other species (). Some of these putative mimics may play roles in inhibition of apoptotic signaling. Rickettsia RC0370 appears to mimic the P-loop NTPase domain of human NACHT proteins involved in immune signaling and apoptosis. Furthermore, adenylate kinase (AK) in Pseudomonas aeruginosa has been shown to act as a virulence factor regulating external ATP-dependent macrophage cell death,Citation67 and the analysis identified several human-like AKs in a range of pathogenic species that represent novel candidate virulence factors (; Table S1A). Another example of potential disruption of apoptotic signaling involves detected between the Anaplasma protein, APH_0455, and the human protein NFBD1/MDC1, with the two proteins sharing a repetitive motif (QPSTSXDQPXT, see Data File S1 for BLAST alignments). Interestingly, Anaplasma phagocytophilum is known to prevent apoptosis in neutrophils,Citation72 and MDC1 is known to have anti-apoptotic activity through inhibition of p53 phosphorylation.Citation47
Before pathogens can appropriate host pathways, they must adhere to and invade host cells. This was the strongest detected mimicry function in our data set, as both the top-scoring putative mimics () and enriched functions () relate to the extracellular matrix and its components (specifically, collagens and leucine-rich repeat proteins). Both collagens and LRRs have been implicated in virulence and play a role in host cell adherence and invasion,Citation20,Citation73,Citation74 and are the focus of the following section. Other detected pathogen proteins with possible roles in host cell adhesion include a Bacillus adhesin (BA_0871), which shares a unique repetitive TEKP motif (Data File S1) with human zonadhesin, and Treponema proteins similar to human ankyrins that may interact with the host cytoskeletonCitation52 ().
Extracellular matrix proteins predicted as major targets of molecular mimicry
Collagens
Prediction #1 () and function enrichment #1 and #2 () relate to detected similarities between human collagens and collagen-like proteins in pathogenic bacteria. and highlight collagens as clear outliers in the E-value and bitscore distributions, similar to that of the known mimicry relationship, RalF-GEF.Citation18 Collagen mimicry was also the most abundant pathogen-specific pattern detected, occurring in 7 pathogens and 0 non-pathogens ().
The top scoring predicted collagen mimic was spr1403 (PclA) from Streptococcus pneumoniae, a protein that has previously been shown to contribute to host-cell adherence and invasion.Citation73
Numerous detected proteins may play similar roles in pathogenesis, including: BA_3841, BCE_1581, BC_3345, CPE0955, CPR_1027, ECs1228, EF_2090, SPy1983, SpyM3_0738, Z1483, lpl2569, BC_2381, CPF_1202, BCE_3739, ECs2941, and Z2340. Another putative collagen mimic detected by the analysis (Spy1983 [SclA] from Streptococcus pyogenes) has recently been demonstrated to act as an adhesin during pathogenesis of streptococcal infection.Citation74 These pathogen-specific collagen-like proteins (CLPs) are significantly more human-like than CLPs found in other bacteria, as shown by the bitscore distributions (). To investigate this further, we analyzed motif content for the predicted subset of collagen mimics found in pathogens, vs. all other CLPs including those found in non-pathogens. The human-like, pathogen-specific subset of CLPs (putative collagen mimics) were found to have significantly more tetrapeptides in common with human collagens than non-pathogen CLPs (Fig. S2). Many of these tetrapeptides contain “GP” motifs characteristic of human collagen sequences. For example, the average number of GP motifs per sequence length is 4.95% for the putative collagen mimics, 4.86% for human collagens, and only 0.26% for predicted non-mimics. Thus, the detected sequence similarities are due to similarities in peptide composition rather than sequence or statistical artifacts.
Leucine-rich repeat proteins
In mammals, leucine-rich repeat proteins are a second class of proteins abundant in the extracellular matrix, where they function in cell growth, adhesion, migration, and bind with other ECM components including collagens.Citation75 As mentioned above, prediction #3 involves detected similarity between human LRRs and internalin-related factors (e.g., lmo0801, LMOh7858_0295), which are known to function in adherence and invasion of host cells (; Table S1A). LRR-containing proteins such as NOD-like receptors and Toll-like receptors also serve as important pathogen-detection molecules, recognizing key pathogen-associated molecular patterns (PAMPs).Citation20,Citation76 Both Toll-like receptors (prediction #27) and NOD-like receptors (prediction #50 and #60) were detected as potential targets of LRR mimicry (Table S1C).
Evolution of ECM mimics via independent repeat events
As the top scoring pattern overall, we analyzed the detected similarities between human extracellular matrix proteins and putative bacterial pathogen mimics. The two detected relationships are largely distinct according to their taxonomic distribution. Detected collagen mimics were associated predominantly with Firmicutes pathogens, and LRR mimics were identified in pathogens from a range of phyla also including Spirochaetes and Bacteroidetes. One species (Streptococcus pyogenes) appears to encode both types of mimicry proteins.
To investigate how the set of putative collagen mimics have evolved in pathogenic bacteria, we analyzed and compared the sequence architecture of predicted ECM mimics from different species. Despite possessing a common repetitive collagen architecture, we found that collagen-like repeats from different bacterial pathogenic species have distinct repeat architectures, indicative of separate evolutionary origins. To demonstrate this, CLPs from different pathogens were divided into their peptide repeats, which were aligned and used to create sequence logos () and phylogenetic trees (, left). While the collagen-like repetitive pattern (GXXN) is common to all detected CLPs, the progenitor repeat sequences are different, and the repeat lengths are also variable (). Phylogenetic trees were constructed based on an alignment of pathogen collagen-like repeats as well as a top-aligning human repeat to the set of repeats within each pathogen protein (see Methods). As revealed by the sequence logos and the tree, the detected collagen mimics from Streptococcus pneumoniae (spr1403), Streptococcus pyogenes (SpyM3_0738), Clostridium perfringens (CPR_1027), Bacillus anthracis (BA_3841), and Legionella pneumophila (lpl2569) appear to have independently evolved their similarity to human collagen via separate repeat amplifications ( and , left). Moreover, different human repeats can be found that cluster specifically with each pathogen repeat class (, left), indicating that different pathogen repeats may not only be mimicking different human proteins, but may be derived from different host peptides.
Figure 4. Independent evolution of ECM mimics from separate repeat amplifications. High-scoring collagen-like (A) and leucine-rich repeat (B) protein mimics were selected and divided into their constituent protein repeats, which were aligned and used to generate sequence logos. Differences between the sequence logos of each repetitive protein suggest evolution from separate progenitor peptides and repeat amplifications. (C) An example demonstrating similarity of leucine-rich repeat sequence conservation patterns between a human NOD-like receptor (NLRC3) and a predicted mimicry candidate (lpl1579) from Legionella pneumophila. The detected level of sequence similarity between these two proteins is far above that observed in non-pathogens (blue) and other pathogens (red) as indicated by the BLAST bitscore distribution (left panel).
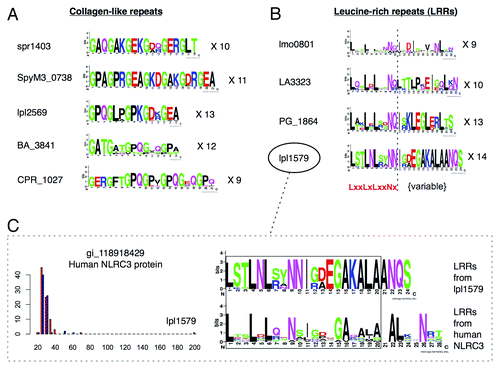
Figure 5. Phylogenetic trees of bacterial pathogen encoded collagen-like repeats (left) and leucine-rich repeats (right) from . The repeats are colored in the tree according to their parent protein. Top-aligning repeats from human proteins have also been included and are colored light green. Repeats cluster predominantly by protein of origin, suggesting that different pathogen repeat proteins have evolved by independent repeat amplifications. Interestingly, the pathogen repeat classes generally cluster with a specific human repeat, suggesting that ancestral progenitor repeats may be host-derived.
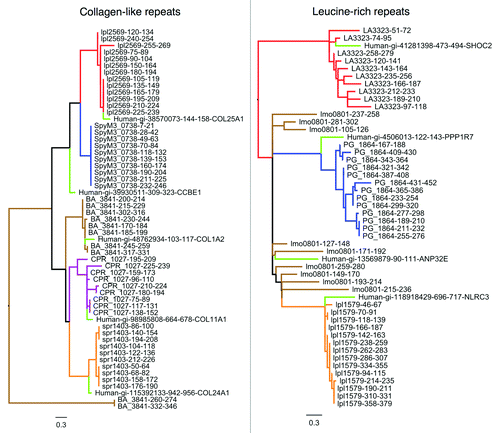
Consistent with the idea that different pathogen CLPs have evolved independently from each other, CLPs in bacteria appear to exhibit a scattered phylogenetic distribution. For example, while CLPs were predominantly detected in pathogens from the Firmicutes phylum, they also exist in the highly pathogenic O157:H7 strain of Escherichia coli (Table S1A) as well as Legionella pneumophila but not other Gammaproteobacteria. Interestingly, while present in E. coli O157:H7, the collagen-like proteins are absent in the uropathogenic E. coli CFT073 strain, and the non-pathogenic K1 strain.
As with the putative collagen mimics, we then analyzed the repeat architecture of the detected LRR mimics, and aligned the sequence logos in the form of the general LRR sequence pattern (based on Ward et al.Citation77) (), and generated phylogenetic trees as described above (, right). Like the detected collagen mimics, LRR mimics from different pathogens exhibit different repeat architectures () and form unique clusters in the phylogenetic tree (, right), implying separate origins via independent repeat amplifications. As described in Ward et al.,Citation77 the first residues LxxLxLxxNx of leucine-rich repeats correspond to a conserved, interior portion of the LRR structure, while the remaining sequence encodes variable residues on the other face of the LRR structure. Interestingly, variable segments from pathogenic LRRs aligned strongly with variable segments from human LRRs, which suggests this may be a variable interaction surface exploited by pathogens. For example, the Legionella protein lpl1579 possesses the motif GAKALA in this variable region, which is similar to the sequence conservation pattern of leucine-rich repeats in human NOD-like receptors (e.g., NLRC3) (). Interestingly, NLRC3 was the top BLAST match to lpl1579 in the human proteome, and it has been demonstrated to have inhibitory effects on T-cell function.Citation78 It is important to note that a BLAST search of lpl1579 across all eukaryotes identified predicted proteins from Naegleria, followed by other NLRC3 proteins from a range of mammals, and so the human NLRC3 is not necessarily the highest scoring hit, which is a general statement that also applies to other detected mimicry relationships. Further phylogenetic analysis would be needed to characterize the evolutionary origins of each detected mimic to verify possible eukaryotic host-bacteria horizontal transferCitation79 from human or non-human host species.
Repetitive and non-repetitive mimics have different functions and modes of evolution
The cases described above (putative collagen and LRR mimics) appear to have evolved independently in pathogens to mimic the repetitive architecture of host proteins.
To investigate the sequence repetition of predicted mimics quantitatively, each region of the putative mimics with detected similarity to human proteins was analyzed and divided into putative sequence repeats using the RADAR repeat prediction algorithm.Citation80 Repeat proteins were found to make up a considerable number of detected mimicry relationships, as 34/95 and 207/306 detected mimics were identified as repetitive (containing three or more predicted repeats) in the non-redundant and full set of predictions, respectively (Table S1).
As revealed by gene-enrichment analyses performed on both sets separately (Table S2B and C), the repetitive class consisting of CLPs, LRRs, and other candidates, are responsible for the enrichments related to extracellular matrix mimicry and cell adhesion (Table S2B). These terms were not significantly enriched among the non-repetitive class (Table S2C). Conversely, non-repetitive mimics were significantly enriched in terms related to enzymatic function such as “catalytic activity” and “lipid-metabolism”, but these were not enriched among the repetitive-class. These results, combined with sequence () and phylogenetic analyses (), are consistent with the idea that non-repetitive mimics with complex sequence composition are associated with enzymatic modulation of host functions and have likely been acquired in pathogens by horizontal transfer, while repetitive mimics have evolved independently in pathogens to mimic repetitive host structural proteins involving in adherence and invasion of host cells.
Another interesting example of the latter is a detected potential mimicry relationship between human protein periaxin and mycobacterial PPE family virulence factors (Rv1918c) (). Mycobacteria such as M. leprae specifically invade human Schwann cells through an interaction with the dystroglycan complex.Citation56,Citation58 Interestingly, human periaxin is a Schwann cell-specific protein that is critical to formation of the dystroglycan-complex. However, as with LRRs and collagens, the detected similarity is not due to homology but rather, a repetitive proline-rich composition that has independently evolved in both proteins (see Data File S1). It is possible that the repetitive proline-rich architecture of PPE proteins may facilitate an interaction with host dystroglycan-complex as is the case for periaxin, or that they may act as membrane-interacting lipoproteins.
Discussion
The results of this work suggest that comparison of host–bacteria proteome similarities is sufficient to detect a subset of pathogen mimics that function in bacterial virulence. Our approach identified known examples of mimicry, virulence factors, and potential novel candidates with roles in modulation and exploitation of host functions.
According to a recent study,Citation81 all human proteins possess motifs also present in bacterial proteins. The same study also found no observable difference in overall bacteria–human peptide similarities between pathogenic and non-pathogenic species. Our results show that while overall sequence similarity to human proteins is not significantly enriched in pathogenic vs. non-pathogenic bacteria, there are detectable pathogen-specific or pathogen-enriched similarities to host proteins in key functional pathways related to virulence. These identified pathways and components, including the extracellular matrix, lipid metabolism, and immune signaling, are known targets of exploitation by bacterial pathogens.
Evolutionary origins of mimicry proteins in pathogenic bacteria
As discussed in previous literature,Citation7,Citation14 two evolutionary mechanisms are likely responsible for detected sequence mimicry between pathogen and host proteins: direct homology due to lateral transfer of the eukaryotic proteins to one or few bacteria, or similarities due to independent evolutionary processes (convergent or parallel evolution) (Fig. S3). In this work, we identified two sequence classes of detected mimics (non-repetitive and repetitive), which likely fall into these two evolutionary categories.
A new insight revealed by this work relates to the independent evolutionary processes by which pathogen mimics can originate. One documented mechanism underlying convergent evolution of host mimicry is independent origin of a binding surfaces or motif in a pathogen protein that displays no detectable homology with its host counterpart.Citation7,Citation14 Our work provides strong support for an additional mechanism, mimicry of host repetitive proteins via independently evolved peptide repeats. In this scenario, separate progenitor repeats in the pathogen genome are amplified to result in repeat proteins that share the same repetitive architecture but with different sequences for each repeat unit (; Fig. S3). This is similar to what has been observed for β-trefoil proteins, which also include virulence-associated subfamilies (i.e., ricin toxins) that have undergone separate repeat amplifications while maintaining the same overall structure.Citation82
These detected similarities do not imply overall homology between the full proteins but rather are due to similarity of repetitive architecture. Repetitive host proteins such as collagens, leucine-rich repeat proteins, and adhesins represent ideal targets for this evolutionary mechanism of pathogen mimicry, while complex proteins such as enzymes are not.
Interestingly, not only do the human and pathogen counterparts of these proteins appear to have evolved independently, but repeat amplifications appear to have occurred independently in different pathogenic species. While this may be indicative of convergent evolution, it is also possible that the pathogen proteins evolved by tandem duplications of an original peptide fragment that itself was acquired from (and thus related to) a host fragment, but then evolved a novel composition through independent evolutionary processes. This is similar in some respects to the results of a recent study that identified a recurring phenomenon whereby host-derived proteins in viruses had subsequently converged toward simpler domain architectures.Citation83
In either case, the enrichment of repetitive ECM mimics in pathogens is likely due to convergent or parallel evolutionary processes that are driven by pathogen-specific selective pressures.
These predictions provide starting points for future experimental work characterizing the biological role of predicted pathogen mimics. We have analyzed only a subset of this spectrum, and future work expanding this analysis, and also evaluating the host-species specificity of this approach will be useful. Thus, future use of alternative classification schemes, improved motif detection techniques and structural bioinformatics may provide added sensitivity.
For instance, the classification scheme that is the basis of our comparative approach separates human pathogenic vs. non-pathogenic bacteria. Although this classification is somewhat arbitrary, the putative mimics detected using this scheme likely play virulence associated roles, and it was an objective of this analysis to find such pathogen-associated mimics. However, it is also important to note that other classes of mimics exist that may not be detected by our pathogen/non-pathogen comparison. These include mimics that are not directly involved in virulence but might still play a role in persistence of commensal bacteria inside the host, or have other effects such as the extensively studied role of peptide mimicry in generation of autoimmune disease.Citation1,Citation2 In the case of immune epitope mimicry, small regions of sequence similarity and not overall homology may be sufficient to elicit molecular mimicry,Citation3,Citation5 which would also not be identified using a standard homology detection approach. Our work thus complements previous work,Citation5 which has focused on such cases of immune epitope mimicry. Finally, pathogen mimics of host proteins may have diverged beyond the point of recognizable sequence homology, but be detectable at the level of overall structural similarity.
Ultimately, to extend computational analysis of host–pathogen molecular mimicry, it may be useful to analyze mimicry with respect to the specific pathology of each bacterial species and the biological consequences they have on their host (e.g., autoimmunity, direct damage, interference of metabolism, persistence in host), as well as conduct sequence comparisons at the level of motif fragments, perhaps taking into account protein structural information. With such improvements, it will be increasingly possible to predict novel virulence mechanisms and host-pathogen relationships from genomic data. A resource containing predicted mimicry candidates discussed in this paper is available at http://doxey.uwaterloo.ca/mimicry/.
Methods
Computational screen for mimicry candidates using all-by-all BLAST analysis
Protein sequence data sets for human as well as 163 bacterial genomes (see Table S1 for a complete list) were retrieved from the NCBI (RefSeq human protein database build 36 [37 742 proteins]) and the Comprehensive Microbial ResourceCitation23 at TIGR/JVCI (http://cmr.jcvi.org). To reduce species redundancy, only one proteome per species was kept and the rest were removed. This step removed 35 species, leaving 62 pathogens and 66 non-pathogens.
An all-by-all BLAST analysis was conducted using BLAST v. 2.2.16, in which each human protein was used as an individual query in a separate BLAST search of each individual organism’s protein database. Default BLAST parameters were used with “composition-based statistics” to correct for potential compositional bias. A BLAST E-value cutoff of 1e−06 was used to identify putative matches, from which a presence/absence matrix was constructed. BLAST E-values, bitscores, and top pathogen protein matches were recorded for each cell of the matrix. To remove genome/species redundancy, only one genome per species was kept (randomly assigned) and the remaining genomes were removed.
Each human protein (i) was then scored using the fraction of pathogen species with a hit detected by blast (Pi) divided by the fraction of non-pathogens (NPi) containing a hit. Potential protein mimics were selected based on rarity in non-pathogens, enrichment in pathogens, and greater similarity between pathogen and human proteins. The specific criteria were hits found in less than five non-pathogens, Pi/NPi ratio greater than 2, and top pathogen BLAST hit had a bitscore greater than 10 above that of the top non-pathogen hit. A bitscore difference of 10 was chosen to result in a final list equivalent to roughly 1% of the human proteome (99% percentile).
The full list of detected mimicry candidates is shown in Table S1. A smaller list of the most unique relationships was also generated by including only the top human matches to each unique pathogen protein. This generated a smaller set of 95 unique mimicry relationships (Table S1C), the top 25 of which are listed in .
Phytopathogen mimicry analysis
As a control, and test for generality, we applied the approach to a different host-pathogen relationship (plant/phytopathogen). The same approach was used as described above with Arabidopsis thaliana used as the host proteome, and the following species were defined as phytopathogens: Phytoplasma asteris, Agrobacterium tumefaciens, Ralstonia solanacearum, Pseudomonas syringae, Xanthomonas axonopodis, and Xylella fastidiosa. Xanthomonas campestris was removed due to redundancy with X. axonopodis. In total, this data set contained six phytopathogens and 16 non-pathogens.
Homology to known virulence factors
All candidate pathogen mimics were searched against the MvirDBCitation27 database of known virulence factors. Hits were again defined as BLAST matches with E < 1e−06.
Gene enrichment analysis
Gene enrichment analysis for overrepresented functions was performed using DAVID.Citation29 The following eight ontologies were used: GOTERM_BP_ALL, GO_TERM_CC_ALL, GOTERM_MF_ALL, PANTHER_BP_ALL, PANTHER_MF_ALL, BIOCARTA, KEGG_PATHWAY, and PANTHER_PATHWAY. The default parameter value of EASE = 0.1 was used.
Detection and analysis of protein repeat sequences
For each putative mimic, the BLAST result was parsed to extract the sequence region detected as similar to the host protein. Repeats were predicted for these regions using RADARCitation80 run with default parameters. Repetitive mimics were defined as those containing greater than three predicted repeats, all of which had total scores > 90.
For the detected collagen-like and LRR mimics, the detected repeats were aligned, adjusted where necessary to a common length, and sequence logos were generated using seqlogo with default parameters (http://weblogo.berkeley.edu).
For each set of repeats from a pathogen protein, the consensus sequence was used as a query to identify the best-aligning repeat from a corresponding human protein using SSEARCH.Citation84 Collagen-like and leucine-rich repeats were then aligned separately along with their respective human sequences and a phylogenetic tree was generated using parsimony using Phylip.Citation85 Branch lengths were estimated by maximum likelihood using FasttreeCitation86 with the JTT model and CAT approximation with 20 rate categories.
Sequence composition analysis
The compseq program within the EMBOSS suiteCitation87 (version 6.3.1) was used to compute motif frequencies for human collagens, the putative collagen mimics and non-mimics.
Additional material
Download Zip (1.7 MB)Acknowledgments
This work was supported by the National Science and Engineering Research Council of Canada (NSERC) through grants to BJM (NSERC Discovery Grant) and ACD (NSERC PDF). We thank Trevor Charles for insightful discussions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Albert LJ, Inman RD. Molecular mimicry and autoimmunity. N Engl J Med 1999; 341:2068 - 74; http://dx.doi.org/10.1056/NEJM199912303412707; PMID: 10615080
- Oldstone MB. Molecular mimicry, microbial infection, and autoimmune disease: evolution of the concept. Curr Top Microbiol Immunol 2005; 296:1 - 17; http://dx.doi.org/10.1007/3-540-30791-5_1; PMID: 16329189
- Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol 2012; 42:102 - 11; http://dx.doi.org/10.1007/s12016-011-8294-7; PMID: 22095454
- Cunningham MW. Streptococcus and rheumatic fever. Curr Opin Rheumatol 2012; 24:408 - 16; http://dx.doi.org/10.1097/BOR.0b013e32835461d3; PMID: 22617826
- Babu Chodisetti S, Rai PK, Gowthaman U, Pahari S, Agrewala JN. Potential T cell epitopes of Mycobacterium tuberculosis that can instigate molecular mimicry against host: implications in autoimmune pathogenesis. BMC Immunol 2012; 13:13; http://dx.doi.org/10.1186/1471-2172-13-13; PMID: 22435930
- Finlay BB, Cossart P. Exploitation of mammalian host cell functions by bacterial pathogens. Science 1997; 276:718 - 25; http://dx.doi.org/10.1126/science.276.5313.718; PMID: 9115192
- Stebbins CE, Galán JE. Structural mimicry in bacterial virulence. Nature 2001; 412:701 - 5; http://dx.doi.org/10.1038/35089000; PMID: 11507631
- Knodler LA, Celli J, Finlay BB. Pathogenic trickery: deception of host cell processes. Nat Rev Mol Cell Biol 2001; 2:578 - 88; http://dx.doi.org/10.1038/35085062; PMID: 11483991
- Bhavsar AP, Guttman JA, Finlay BB. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007; 449:827 - 34; http://dx.doi.org/10.1038/nature06247; PMID: 17943119
- Elde NC, Malik HS. The evolutionary conundrum of pathogen mimicry. Nat Rev Microbiol 2009; 7:787 - 97; http://dx.doi.org/10.1038/nrmicro2222; PMID: 19806153
- Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol 2001; 55:709 - 42; http://dx.doi.org/10.1146/annurev.micro.55.1.709; PMID: 11544372
- Gilk SD, Beare PA, Heinzen RA. Coxiella burnetii expresses a functional Δ24 sterol reductase. J Bacteriol 2010; 192:6154 - 9; http://dx.doi.org/10.1128/JB.00818-10; PMID: 20870767
- Omsland A, Heinzen RA. Life on the outside: the rescue of Coxiella burnetii from its host cell. Annu Rev Microbiol 2011; 65:111 - 28; http://dx.doi.org/10.1146/annurev-micro-090110-102927; PMID: 21639786
- Sikora S, Strongin A, Godzik A. Convergent evolution as a mechanism for pathogenic adaptation. Trends Microbiol 2005; 13:522 - 7; http://dx.doi.org/10.1016/j.tim.2005.08.010; PMID: 16153847
- Sallee NA, Rivera GM, Dueber JE, Vasilescu D, Mullins RD, Mayer BJ, et al. The pathogen protein EspF(U) hijacks actin polymerization using mimicry and multivalency. Nature 2008; 454:1005 - 8; http://dx.doi.org/10.1038/nature07170; PMID: 18650806
- Hamburger ZA, Brown MS, Isberg RR, Bjorkman PJ. Crystal structure of invasin: a bacterial integrin-binding protein. Science 1999; 286:291 - 5; http://dx.doi.org/10.1126/science.286.5438.291; PMID: 10514372
- Graham SC, Bahar MW, Cooray S, Chen RA, Whalen DM, Abrescia NG, et al. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-kappaB rather than apoptosis. PLoS Pathog 2008; 4:e1000128; http://dx.doi.org/10.1371/journal.ppat.1000128; PMID: 18704168
- Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 2002; 295:679 - 82; http://dx.doi.org/10.1126/science.1067025; PMID: 11809974
- Marino M, Braun L, Cossart P, Ghosh P. Structure of the lnlB leucine-rich repeats, a domain that triggers host cell invasion by the bacterial pathogen L. monocytogenes. Mol Cell 1999; 4:1063 - 72; http://dx.doi.org/10.1016/S1097-2765(00)80234-8; PMID: 10635330
- Kedzierski Ł, Montgomery J, Curtis J, Handman E. Leucine-rich repeats in host-pathogen interactions. Arch Immunol Ther Exp (Warsz) 2004; 52:104 - 12; PMID: 15179324
- Doxey AC, Lynch MD, Müller KM, Meiering EM, McConkey BJ. Insights into the evolutionary origins of clostridial neurotoxins from analysis of the Clostridium botulinum strain A neurotoxin gene cluster. BMC Evol Biol 2008; 8:316; http://dx.doi.org/10.1186/1471-2148-8-316; PMID: 19014598
- Ludin P, Nilsson D, Mäser P. Genome-wide identification of molecular mimicry candidates in parasites. PLoS One 2011; 6:e17546; http://dx.doi.org/10.1371/journal.pone.0017546; PMID: 21408160
- Peterson JD, Umayam LA, Dickinson T, Hickey EK, White O. The Comprehensive Microbial Resource. Nucleic Acids Res 2001; 29:123 - 5; http://dx.doi.org/10.1093/nar/29.1.123; PMID: 11125067
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25:3389 - 402; http://dx.doi.org/10.1093/nar/25.17.3389; PMID: 9254694
- Moran NA. Microbial minimalism: genome reduction in bacterial pathogens. Cell 2002; 108:583 - 6; http://dx.doi.org/10.1016/S0092-8674(02)00665-7; PMID: 11893328
- Orchard RC, Alto NM. Mimicking GEFs: a common theme for bacterial pathogens. Cell Microbiol 2012; 14:10 - 8; http://dx.doi.org/10.1111/j.1462-5822.2011.01703.x; PMID: 21951829
- Zhou CE, Smith J, Lam M, Zemla A, Dyer MD, Slezak T. MvirDB--a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res 2007; 35:Database issue D391 - 4; http://dx.doi.org/10.1093/nar/gkl791; PMID: 17090593
- Sun HY, Lin SW, Ko TP, Pan JF, Liu CL, Lin CN, et al. Structure and mechanism of Helicobacter pylori fucosyltransferase. A basis for lipopolysaccharide variation and inhibitor design. J Biol Chem 2007; 282:9973 - 82; http://dx.doi.org/10.1074/jbc.M610285200; PMID: 17251184
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44 - 57; http://dx.doi.org/10.1038/nprot.2008.211; PMID: 19131956
- Weinrauch Y, Zychlinsky A. The induction of apoptosis by bacterial pathogens. Annu Rev Microbiol 1999; 53:155 - 87; http://dx.doi.org/10.1146/annurev.micro.53.1.155; PMID: 10547689
- Lukomski S, Nakashima K, Abdi I, Cipriano VJ, Ireland RM, Reid SD, et al. Identification and characterization of the scl gene encoding a group A Streptococcus extracellular protein virulence factor with similarity to human collagen. Infect Immun 2000; 68:6542 - 53; http://dx.doi.org/10.1128/IAI.68.12.6542-6553.2000; PMID: 11083763
- Gaillard JL, Berche P, Frehel C, Gouin E, Cossart P. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 1991; 65:1127 - 41; http://dx.doi.org/10.1016/0092-8674(91)90009-N; PMID: 1905979
- Koonin EV, Aravind L. The NACHT family - a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem Sci 2000; 25:223 - 4; http://dx.doi.org/10.1016/S0968-0004(00)01577-2; PMID: 10782090
- Koonin EV, Aravind L. Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ 2002; 9:394 - 404; http://dx.doi.org/10.1038/sj.cdd.4400991; PMID: 11965492
- Zaborina O, Li X, Cheng G, Kapatral V, Chakrabarty AM. Secretion of ATP-utilizing enzymes, nucleoside diphosphate kinase and ATPase, by Mycobacterium bovis BCG: sequestration of ATP from macrophage P2Z receptors?. Mol Microbiol 1999; 31:1333 - 43; http://dx.doi.org/10.1046/j.1365-2958.1999.01240.x; PMID: 10200955
- Punj V, Zaborina O, Dhiman N, Falzari K, Bagdasarian M, Chakrabarty AM. Phagocytic cell killing mediated by secreted cytotoxic factors of Vibrio cholerae. Infect Immun 2000; 68:4930 - 7; http://dx.doi.org/10.1128/IAI.68.9.4930-4937.2000; PMID: 10948107
- Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal 2009; 2:pe6; http://dx.doi.org/10.1126/scisignal.256pe6; PMID: 19193605
- Chen J, de Felipe KS, Clarke M, Lu H, Anderson OR, Segal G, et al. Legionella effectors that promote nonlytic release from protozoa. Science 2004; 303:1358 - 61; http://dx.doi.org/10.1126/science.1094226; PMID: 14988561
- Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 2007; 450:365 - 9; http://dx.doi.org/10.1038/nature06336; PMID: 17952054
- Wheeler PR. Pyrimidine scavenging by Mycobacterium leprae. FEMS Microbiol Lett 1989; 48:179 - 84; http://dx.doi.org/10.1111/j.1574-6968.1989.tb03295.x; PMID: 2656380
- Zhang JR, Idanpaan-Heikkila I, Fischer W, Tuomanen EI. Pneumococcal licD2 gene is involved in phosphorylcholine metabolism. Mol Microbiol 1999; 31:1477 - 88; http://dx.doi.org/10.1046/j.1365-2958.1999.01291.x; PMID: 10200966
- Deb C, Lee CM, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, et al. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 2009; 4:e6077; http://dx.doi.org/10.1371/journal.pone.0006077; PMID: 19562030
- Ehrt S, Schnappinger D. Mycobacterium tuberculosis virulence: lipids inside and out. Nat Med 2007; 13:284 - 5; http://dx.doi.org/10.1038/nm0307-284; PMID: 17342139
- Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res 1996; 24:4420 - 49; http://dx.doi.org/10.1093/nar/24.22.4420; PMID: 8948633
- Yu Y, Sun G, Liu G, Wang Y, Shao Z, Chen Z, et al. Effects of Mycoplasma pneumoniae infection on sphingolipid metabolism in human lung carcinoma A549 cells. Microb Pathog 2009; 46:63 - 72; http://dx.doi.org/10.1016/j.micpath.2008.10.014; PMID: 19059331
- Smith GA, Marquis H, Jones S, Johnston NC, Portnoy DA, Goldfine H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun 1995; 63:4231 - 7; PMID: 7591052
- Nakanishi M, Ozaki T, Yamamoto H, Hanamoto T, Kikuchi H, Furuya K, et al. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. J Biol Chem 2007; 282:22993 - 3004; http://dx.doi.org/10.1074/jbc.M611412200; PMID: 17535811
- Faherty CS, Maurelli AT. Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol 2008; 16:173 - 80; http://dx.doi.org/10.1016/j.tim.2008.02.001; PMID: 18353648
- Sohlenkamp C, López-Lara IM, Geiger O. Biosynthesis of phosphatidylcholine in bacteria. Prog Lipid Res 2003; 42:115 - 62; http://dx.doi.org/10.1016/S0163-7827(02)00050-4; PMID: 12547654
- Kent C, Gee P, Lee SY, Bian X, Fenno JC. A CDP-choline pathway for phosphatidylcholine biosynthesis in Treponema denticola. Mol Microbiol 2004; 51:471 - 81; http://dx.doi.org/10.1046/j.1365-2958.2003.03839.x; PMID: 14756787
- Ivanova N, Sorokin A, Anderson I, Galleron N, Candelon B, Kapatral V, et al. Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 2003; 423:87 - 91; http://dx.doi.org/10.1038/nature01582; PMID: 12721630
- Weinstock GM, Hardham JM, McLeod MP, Sodergren EJ, Norris SJ. The genome of Treponema pallidum: new light on the agent of syphilis. FEMS Microbiol Rev 1998; 22:323 - 32; http://dx.doi.org/10.1111/j.1574-6976.1998.tb00373.x; PMID: 9862125
- Price CT, Al-Khodor S, Al-Quadan T, Santic M, Habyarimana F, Kalia A, et al. Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitination machinery within macrophages and protozoa. PLoS Pathog 2009; 5:e1000704; http://dx.doi.org/10.1371/journal.ppat.1000704; PMID: 20041211
- Black DS, Bliska JB. Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO J 1997; 16:2730 - 44; http://dx.doi.org/10.1093/emboj/16.10.2730; PMID: 9184219
- Ren SX, Fu G, Jiang XG, Zeng R, Miao YG, Xu H, et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature 2003; 422:888 - 93; http://dx.doi.org/10.1038/nature01597; PMID: 12712204
- Marques MA, Ant nio VL, Sarno EN, Brennan PJ, Pessolani MC. Binding of alpha2-laminins by pathogenic and non-pathogenic mycobacteria and adherence to Schwann cells. J Med Microbiol 2001; 50:23 - 8; PMID: 11192500
- Sherman DL, Fabrizi C, Gillespie CS, Brophy PJ. Specific disruption of a schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron 2001; 30:677 - 87; http://dx.doi.org/10.1016/S0896-6273(01)00327-0; PMID: 11430802
- Rambukkana A, Yamada H, Zanazzi G, Mathus T, Salzer JL, Yurchenco PD, et al. Role of alpha-dystroglycan as a Schwann cell receptor for Mycobacterium leprae. Science 1998; 282:2076 - 9; http://dx.doi.org/10.1126/science.282.5396.2076; PMID: 9851927
- Brüggemann H, Cazalet C, Buchrieser C. Adaptation of Legionella pneumophila to the host environment: role of protein secretion, effectors and eukaryotic-like proteins. Curr Opin Microbiol 2006; 9:86 - 94; http://dx.doi.org/10.1016/j.mib.2005.12.009; PMID: 16406773
- Sansom FM, Newton HJ, Crikis S, Cianciotto NP, Cowan PJ, d’Apice AJ, et al. A bacterial ecto-triphosphate diphosphohydrolase similar to human CD39 is essential for intracellular multiplication of Legionella pneumophila. Cell Microbiol 2007; 9:1922 - 35; http://dx.doi.org/10.1111/j.1462-5822.2007.00924.x; PMID: 17388784
- Xu Y, Liang X, Chen Y, Koehler TM, Höök M. Identification and biochemical characterization of two novel collagen binding MSCRAMMs of Bacillus anthracis. J Biol Chem 2004; 279:51760 - 8; http://dx.doi.org/10.1074/jbc.M406417200; PMID: 15456768
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Pontén T, Alsmark UC, Podowski RM, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998; 396:133 - 40; http://dx.doi.org/10.1038/24094; PMID: 9823893
- Sikora AE, Zielke RA, Lawrence DA, Andrews PC, Sandkvist M. Proteomic analysis of the Vibrio cholerae type II secretome reveals new proteins, including three related serine proteases. J Biol Chem 2011; 286:16555 - 66; http://dx.doi.org/10.1074/jbc.M110.211078; PMID: 21385872
- Walker DH, Feng HM, Popov VL. Rickettsial phospholipase A2 as a pathogenic mechanism in a model of cell injury by typhus and spotted fever group rickettsiae. Am J Trop Med Hyg 2001; 65:936 - 42; PMID: 11792002
- Rahman MS, Ammerman NC, Sears KT, Ceraul SM, Azad AF. Functional characterization of a phospholipase A(2) homolog from Rickettsia typhi. J Bacteriol 2010; 192:3294 - 303; http://dx.doi.org/10.1128/JB.00155-10; PMID: 20435729
- Soccio RE, Breslow JL. StAR-related lipid transfer (START) proteins: mediators of intracellular lipid metabolism. J Biol Chem 2003; 278:22183 - 6; http://dx.doi.org/10.1074/jbc.R300003200; PMID: 12724317
- Markaryan A, Zaborina O, Punj V, Chakrabarty AM. Adenylate kinase as a virulence factor of Pseudomonas aeruginosa. J Bacteriol 2001; 183:3345 - 52; http://dx.doi.org/10.1128/JB.183.11.3345-3352.2001; PMID: 11344142
- Rottem S, Adar L, Gross Z, Ne’eman Z, Davis PJ. Incorporation and modification of exogenous phosphatidylcholines by mycoplasmas. J Bacteriol 1986; 167:299 - 304; PMID: 3087959
- Dixon TC, Fadl AA, Koehler TM, Swanson JA, Hanna PC. Early Bacillus anthracis-macrophage interactions: intracellular survival survival and escape. Cell Microbiol 2000; 2:453 - 63; http://dx.doi.org/10.1046/j.1462-5822.2000.00067.x; PMID: 11207600
- la Sala A, Ferrari D, Di Virgilio F, Idzko M, Norgauer J, Girolomoni G. Alerting and tuning the immune response by extracellular nucleotides. J Leukoc Biol 2003; 73:339 - 43; http://dx.doi.org/10.1189/jlb.0802418; PMID: 12629147
- Zheng LM, Zychlinsky A, Liu CC, Ojcius DM, Young JD. Extracellular ATP as a trigger for apoptosis or programmed cell death. J Cell Biol 1991; 112:279 - 88; http://dx.doi.org/10.1083/jcb.112.2.279; PMID: 1988462
- Yoshiie K, Kim HY, Mott J, Rikihisa Y. Intracellular infection by the human granulocytic ehrlichiosis agent inhibits human neutrophil apoptosis. Infect Immun 2000; 68:1125 - 33; http://dx.doi.org/10.1128/IAI.68.3.1125-1133.2000; PMID: 10678916
- Paterson GK, Nieminen L, Jefferies JM, Mitchell TJ. PclA, a pneumococcal collagen-like protein with selected strain distribution, contributes to adherence and invasion of host cells. FEMS Microbiol Lett 2008; 285:170 - 6; http://dx.doi.org/10.1111/j.1574-6968.2008.01217.x; PMID: 18557785
- Chen SM, Tsai YS, Wu CM, Liao SK, Wu LC, Chang CS, et al. Streptococcal collagen-like surface protein 1 promotes adhesion to the respiratory epithelial cell. BMC Microbiol 2010; 10:320; http://dx.doi.org/10.1186/1471-2180-10-320; PMID: 21159159
- Hocking AM, Shinomura T, McQuillan DJ. Leucine-rich repeat glycoproteins of the extracellular matrix. Matrix Biol 1998; 17:1 - 19; http://dx.doi.org/10.1016/S0945-053X(98)90121-4; PMID: 9628249
- Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol 2006; 7:1250 - 7; http://dx.doi.org/10.1038/ni1412; PMID: 17110941
- Ward CW, Garrett TP. The relationship between the L1 and L2 domains of the insulin and epidermal growth factor receptors and leucine-rich repeat modules. BMC Bioinformatics 2001; 2:4; http://dx.doi.org/10.1186/1471-2105-2-4; PMID: 11504559
- Conti BJ, Davis BK, Zhang J, O’connor W Jr., Williams KL, Ting JP. CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J Biol Chem 2005; 280:18375 - 85; http://dx.doi.org/10.1074/jbc.M413169200; PMID: 15705585
- Böttger A, Doxey AC, Hess MW, Pfaller K, Salvenmoser W, Deutzmann R, et al. Horizontal gene transfer contributed to the evolution of extracellular surface structures: the freshwater polyp Hydra is covered by a complex fibrous cuticle containing glycosaminoglycans and proteins of the PPOD and SWT (sweet tooth) families. PLoS One 2012; 7:e52278; http://dx.doi.org/10.1371/journal.pone.0052278; PMID: 23300632
- Heger A, Holm L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins 2000; 41:224 - 37; http://dx.doi.org/10.1002/1097-0134(20001101)41:2<224::AID-PROT70>3.0.CO;2-Z; PMID: 10966575
- Trost B, Lucchese G, Stufano A, Bickis M, Kusalik A, Kanduc D. No human protein is exempt from bacterial motifs, not even one. Self Nonself 2010; 1:328 - 34; http://dx.doi.org/10.4161/self.1.4.13315; PMID: 21487508
- Broom A, Doxey AC, Lobsanov YD, Berthin LG, Rose DR, Howell PL, et al. Modular evolution and the origins of symmetry: reconstruction of a three-fold symmetric globular protein. Structure 2012; 20:161 - 71; http://dx.doi.org/10.1016/j.str.2011.10.021; PMID: 22178248
- Rappoport N, Linial M. Viral proteins acquired from a host converge to simplified domain architectures. PLoS Comput Biol 2012; 8:e1002364; http://dx.doi.org/10.1371/journal.pcbi.1002364; PMID: 22319434
- Pearson WR. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics 1991; 11:635 - 50; http://dx.doi.org/10.1016/0888-7543(91)90071-L; PMID: 1774068
- Felsenstein J. PHYLIP–Phylogeny Inference Package (Version 3.2). Cladistics 1981; 5:164 - 6
- Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 2010; 5:e9490; http://dx.doi.org/10.1371/journal.pone.0009490; PMID: 20224823
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 2000; 16:276 - 7; http://dx.doi.org/10.1016/S0168-9525(00)02024-2; PMID: 10827456