Abstract
Bacterial infections, like their viral counterparts, trigger the onset of innate immune defense mechanisms through the release of cytokines, including interferons (IFNs). While type I and II IFN responses to bacteria have long been explored, type III IFN response remains poorly addressed. We have recently reported that the pathogen Listeria monocytogenes triggers the expression of type I and III IFN genes in epithelial cells, and is able to fine-tune downstream signaling at the chromatin level. This bacterium can negatively or positively modulate the expression of interferon-stimulated genes (ISGs) by manipulating the function of BAHD1, a component of a host chromatin-silencing complex. To this end, L. monocytogenes tightly controls the secretion of a BAHD1 inhibitory factor, LntA. Here, we further document the current knowledge about chromatin mechanisms modulating interferon responses during host-bacteria interplay, and discuss their physiological consequences.
When intracellular bacteria infect eukaryotic cells, a number of signaling events lead to a drastic reprogramming of the host cell transcriptional landscape. A growing body of evidence indicates that these local or genome-wide changes can proceed from bacterial-induced chromatin modifications.Citation1,Citation2 Most of the reported mechanisms of chromatin subversion by bacteria result in the silencing of key host defense genes. For instance, the secreted protein phosphatase OspF from Shigella flexneri prevents the phosphorylation of histone H3 on serine 10 (H3S10) at the promoters of a set of innate immune genes including IL-8 and consequently downregulates these genes.Citation3 Exposure of HeLa cells to bacterial pore-formin toxins, such as Listeria monocytogenes listeriolysin O (LLO), also results in the rapid dephosphorylation of H3S10 at the promoters of various host genes, including the innate immune-related genes CXCL2 and IFIT3, and correlates with their downregulation.Citation4 However, gene silencing does not only depend on histone modifications; this multi-step process requires the coordinated action of DNA-, RNA- and histone-binding, -modifying and -remodeling factors. These components assemble into large complexes, establishing a condensed chromatin state referred to as heterochromatin and leading to repression of transcription. We focus here on the subversion by bacteria of some of these chromatin regulators in the context of interferon responses.
Two Bacteria Can Manipulate Chromatin to Regulate Interferon Responses
Interferons are major cytokines produced in response to both viral and bacterial infection.Citation5 After recognition of pathogen determinants, type II IFNs (IFN-γ) are produced exclusively by immune cells, while types I and III (IFN-I and IFN-III) can be produced by a wide range of cell types. Upon binding to their cognate surface receptor, they activate JAK-STAT (Janus Kinase/Signal Transducer and Activator of Transcription)-signaling pathways. Typically, IFN-γ will trigger the translocation of phosphorylated STAT1/STAT1 homodimers to the nucleus, and induce the transcription of a subset of genes characterized by the presence of a GAS (gamma-interferon activated site) box in their promoter sequence. In contrast, IFN-Ι and IFN-III stimulations predominantly induce the formation of a phosphorylated STAT1/STAT2/IRF9 heterotrimer, also named ISGF3 (ISG Factor 3). After entering the nucleus, this complex binds to promoters containing Interferon-Sensitive Response Elements (ISREs) and drives the transcription of downstream genes.
Mycobacterium tuberculosis and M. avium were the first bacteria reported to control interferon-induced pathways at the chromatin level.Citation6-Citation8 During infection of macrophages stimulated with IFN-γ, mycobacteria repress some IFN-γ-responsive genes, in particular genes encoding major histocompatibility complex class II (MHC-II) molecules and their transactivator CIITA (). The recognition of a mycobacterial cell wall lipoprotein, LpqH, by TLR2, and subsequent signaling through the MAPK pathways, result in the binding of the transcription factor C/EBP at the promoter of CIITA. This prevents the recruitment of the SWI/SNF chromatin remodeler at this site and leads to histone deacetylation and CIITA repression.Citation7 CIITA-regulated genes, such as HLA-DR, are (1) downregulated by the decrease in CIITA amounts and (2) further repressed by the recruitment at their promoter of a chromatin complex containing histone deacetylases (HDACs) and the co-repressor mSin3A, in response to TLR2 activation.Citation8
Figure 1. Bacterial modulation of interferon responses by chromatin-related mechanisms. In macrophages, the activation of TLR2 by Mycobacterium counteracts IFN-II-dependent signaling. Activation of MAPKs triggers the activation of histone deacetylases and (1) the silencing of the CIITA gene, (2) the repression of CIITA-dependent gene expression. In epithelial cells, infection by Listeria leads to the expression of IFN-I and IFN-III genes (not represented). Subsequently, these cytokines activate the JAK/STAT pathway. However, by an unknown pathway, infection also drives the BAHD1-associated chromatin complex to repress ISGs. When Listeria secretes LntA, this factor enters the nucleus where it interacts with BAHD1, destabilizes the silencing complex, restores H3K9 acetylation (Ac) and enhances the expression of ISGs.
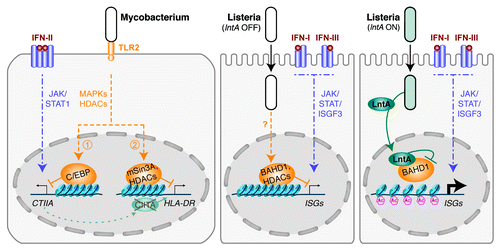
Listeria monocytogenes is the second example of a bacterium controlling the expression of ISGs by acting on the chromatin remodeling machinery.Citation9 In contrast to mycobacteria, Listeria chromatin modulation targets IFN-I and IFN-III pathways in epithelial cells (). Our recent work suggests that infection by Listeria promotes the repressive function of a chromatin-silencing complex at the promoter of a set of ISRE-dependent ISGs. We established that two components of this complex, the heterochromatinization factor BAHD1Citation10 and the co-repressor KAP1, participate in the downregulation of ISGs. Further studies will be needed to investigate the signaling pathways triggering this bacterial-mediated repression of ISGs, as well as the role of BAHD1/KAP1 partners ().
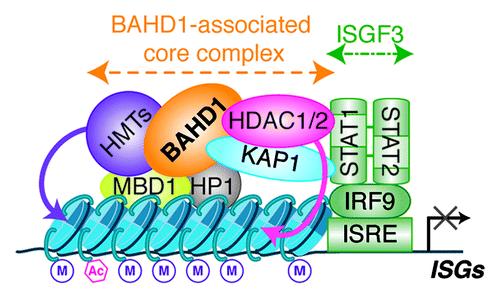
Figure 2. Model of the BAHD1-associated repressive core complex deposited at the promoters of ISGs. The BAHD1-associated chromatin complex contains several heterochromatin components, such as KAP1, HP1 and MBD1, and modifying enzymes, such as histone methyltransferases (HMTs) and deacetylases (HDACs). Since KAP1 and HDAC1 can interact with STAT proteins, we hypothesize that they could bridge the silencing complex with ISGF3, deposited on ISRE sequences. HP1 was also described as a regulator of ISGs. HMTs can methylate histones (M), while HDACs can erase acetylations (Ac), resulting in a closed chromatin state at targeted promoters.
Remarkably, Listeria has evolved a strategy to counterbalance ISG repression in specific conditions, by directly targeting this chromatin complex. A secreted virulence factor, LntA, enters the nucleus of infected cells, where it interacts with BAHD1, impedes its function, and thereby promotes the expression of ISGs. We observed a decrease in BAHD1 occupancy of the promoters of IFITM1 and IFIT3 in cells infected with LntA-producing bacteria. Consistent with the presence of HDAC1/2 in the BAHD1-associated complex, the level of acetylation of lysine 9 on histone H3, which is a mark of active chromatin, increased at the promoters of ISGs in the presence of LntA. How LntA targets BAHD1 specifically at ISGs remains an open question. Indeed, LntA had no effect on either BAHD1 occupancy or histone acetylation status at the promoter of IGF2, one of the genes that BAHD1 represses constitutively.Citation10 The specificity of LntA recruitment to ISRE-containing promoters might be driven through an interaction with transcription factors activated only in infected cells, such as ISGF3. LntA would thus displace BAHD1 only from a subset of infection-dependent targets.
Chromatin Complexes Regulate Interferon Responses
The above-described studies highlighted that mechanisms controlling interferon responses in infected cells highly depend on chromatin states and allowed the identification of new chromatin regulators, in particular BAHD1.Citation10 Remarkably, some of the components that we identified in the chromatin complex associated to BAHD1 () are general modulators of ISGs in interferon-stimulated cells and not only during bacterial infections. For example, the histone deacetylase HDAC1 has been shown to directly bind STAT1 and 2, and modulate IFN-α-induced transcription.Citation11 The heterochromatin proteins HP1 can repress the transcription of ISGs, through the interaction of their chromoshadow-domain with H3, antagonizing the nucleosome-remodeling activity of the SWI/SNF complex.Citation12 The pleiotropic chromatin regulator KAP1 is able to bind STAT1, 3, 4 and 6 transcription factors,Citation13 and represses type-I interferon-dependent transcription in HeLa cells.Citation14 In addition, BAHD1 interacts with H3K9 methyltransferases such as STDB1/KMT1E and the methylated-DNA binding protein MBD1Citation10; these factors might promote heterochromatin formation at the promoters of ISGs in response to IFN-Ι and IFN-ΙΙΙ signaling, as was shown for MBD1 in response to IFN-γ.Citation15
Beside chromatin-silencing factors, proteins that open the chromatin structure, such as the chromatin-remodeling complex of the SWI/SNF family PBAF, can also regulate the expression of ISGs. In response to IFN-α or viral challenge, the BAF47 subunit of the PBAF complex is required for maintaining an open chromatin state at ISG promoters, thus allowing their induction,Citation16 while the BAF200 subunit confers gene selectivity in regulating ISGs.Citation17
We hypothesize that all these proteins can form distinct regulatory sub-complexes, depending on cell types and stimuli they are submitted to. Specific assortment of factors and targeting of different promoters could thus translate interferon signals into a wide range of cellular responses.
Are Interferon Responses a Blessing or a Threat?
L. monocytogenes uses sophisticated strategies to fine-tune interferon responses, in epithelial cells as discussed here, and in immune cells.Citation5,Citation18 Does this dual capacity to repress or activate innate immune genes translate into a benefit for the pathogen? Most notably, why would a virulence factor be dedicated to the upregulation of ISGs? Here, we emphasize that, while the function of interferons in anti-viral defense mechanisms is widely acknowledged, their role in the context of intracellular bacterial infections is more ambiguous.Citation5,Citation19 Considering the induction of interferon responses only as a line of anti-bacterial defense at the systemic level would constitute a simplistic view.
The production of IFN-γ by macrophages and other immune cells promotes bacterial clearance and is thus critical in controlling primary L. monocytogenes infections.Citation20 In contrast, several independent studies have reported that stimulation of IFN-I production could increase the virulence of intracellular bacterial pathogens, including Listeria, as well as Mycobacterium, Chlamydia and Tropheryma, through various effector mechanisms.Citation5,Citation21,Citation22 In our study, we found that both IFN-I and IFN-III were induced in response to infection of epithelial cells by L. monocytogenes. In the context of viral infections, type III IFNs can substitute for type I in specific tissues, such as epithelia.Citation23 Taken together, it is tempting to speculate that IFN-III might, like IFN-I, have injurious consequences for the host during listeriosis—and perhaps other bacterial infections, although this has not yet been explored in animal models. If so, LntA, by exacerbating the downstream response, would be beneficial to the pathogen, which would explain why ΔlntA mutants are less virulent than wild-type strains. In the future, more detailed in vivo studies will be required to fully understand the contribution of LntA and of its cellular partners on the outcome of Listeria infections. The stimulation of interferon responses might support localized pro-bacterial conditions, such as the recruitment of vehicle immune cells to infected epithelia or the facilitation of bacterial shedding at the intestinal or placental barriers through the destruction of infected tissue or abortion. Otherwise, as was proposed for IFN-I response, the stimulation of ISGs by LntA might inhibit IFN-γ-mediated immunityCitation24 or promote host T-cell apoptosis.Citation25
Nevertheless, the pro-bacterial effect of interferon responses is probably limited to a narrow spatiotemporal frame. Indeed, the constitutive expression of lntA leads to increased bacterial clearance. Consistent with this, BAHD1+/− mice are more resistant to listeriosis than their wild-type littermates. A systemic induction of ISGs during infection thus seems detrimental to bacteria. As a corollary, the expression of lntA appears to be tightly controlled by bacteria. The positive function of LntA, paralleling its production, is thus likely restricted to a specific time and place. We propose that an early and/or acute type I/III interferon response could activate the host innate immune defenses and help fight against Listeria, as it was reported for other bacteria.Citation5,Citation19
ISGs are expressed in distinct combinations depending on the cell type and stimulatory pathways, and they cover a very wide range of cellular or extra-cellular functions. Because physiological effects of interferons are generally studied at the systemic level, functional knowledge concerning individual ISGs remains elusive. An important issue for forthcoming research will thus be to understand more precisely the roles of the ISGs modulated by BAHD1 and LntA. Most of the characterization efforts have so far focused on anti-viral effectors such as the double-stranded RNA-activated serine/threonine protein kinase (PKR) or the 2′-5′-oligo-adenylate synthetase (OAS) gene family, which seem less pertinent to bacterial infections. Functional insights into other ISGs are emerging; for instance, IFI6 (G1P3) and IFI27 (ISG12) are mitochondrial proteins, reported to have respectively anti- and pro-apoptitic effectsCitation26; the ISG15 deconjugating enzyme USP18 and GBP-1 also display anti-apoptotic properties,Citation27,Citation28 while IFIT2 (ISG54) is pro-apoptotic.Citation29 Tuning the balance between various effectors might constitute a way for the bacteria on the one hand to maintain cell survival during intracellular proliferation and on the other hand to trigger cell death, allowing escape from immune response or dissemination.
In conclusion, our recent work has highlighted that intracellular bacteria are able to carefully regulate the response of their host cells to type I and III interferons by targeting directly the cellular chromatin silencing outfit. During infection, this bacteria/host interplay results in modifications of the chromatin status and transcriptional levels of ISGs. Whether these changes will in the long run translate into stable epigenetic marks deserves further investigation.
Acknowledgments
We are grateful to Lilliana Radoshevich for critical reading of the manuscript. Studies in our lab were funded by Institut Pasteur, INRA, INSERM, French Ligue Nationale Contre le Cancer (LNCC RS10/75–76 Bierne), European Research Council (Advanced Grant 233348) and French National Research Agency (ANR, grant EPILIS).
References
- Hamon MA, Cossart P. Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe 2008; 4:100 - 9; http://dx.doi.org/10.1016/j.chom.2008.07.009; PMID: 18692770
- Paschos K, Allday MJ. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol 2010; 18:439 - 47; http://dx.doi.org/10.1016/j.tim.2010.07.003; PMID: 20724161
- Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, et al. An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol 2007; 8:47 - 56; http://dx.doi.org/10.1038/ni1423; PMID: 17159983
- Hamon MA, Batsche E, Regnault B, Tham TN, Seveau S, Muchardt C, et al. Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci USA 2007; 104:13467 - 72; http://dx.doi.org/10.1073/pnas.0702729104; PMID: 17675409
- Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol 2005; 5:675 - 87; http://dx.doi.org/10.1038/nri1684; PMID: 16110316
- Kincaid EZ, Ernst JD. Mycobacterium tuberculosis exerts gene-selective inhibition of transcriptional responses to IFN-gamma without inhibiting STAT1 function. J Immunol 2003; 171:2042 - 9; PMID: 12902509
- Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol 2006; 176:4323 - 30; PMID: 16547269
- Wang Y, Curry HM, Zwilling BS, Lafuse WP. Mycobacteria inhibition of IFN-gamma induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J Immunol 2005; 174:5687 - 94; PMID: 15843570
- Lebreton A, Lakisic G, Job V, Fritsch L, Tham TN, Camejo A, et al. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science 2011; 331:1319 - 21; http://dx.doi.org/10.1126/science.1200120; PMID: 21252314
- Bierne H, Tham TN, Batsche E, Dumay A, Leguillou M, Kerneis-Golsteyn S, et al. Human BAHD1 promotes heterochromatic gene silencing. Proc Natl Acad Sci USA 2009; 106:13826 - 31; http://dx.doi.org/10.1073/pnas.0901259106; PMID: 19666599
- Nusinzon I, Horvath CM. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc Natl Acad Sci USA 2003; 100:14742 - 7; http://dx.doi.org/10.1073/pnas.2433987100; PMID: 14645718
- Lavigne M, Eskeland R, Azebi S, Saint-Andre V, Jang SM, Batsche E, et al. Interaction of HP1 and Brg1/Brm with the globular domain of histone H3 is required for HP1-mediated repression. PLoS Genet 2009; 5:e1000769; http://dx.doi.org/10.1371/journal.pgen.1000769; PMID: 20011120
- Tsuruma R, Ohbayashi N, Kamitani S, Ikeda O, Sato N, Muromoto R, et al. Physical and functional interactions between STAT3 and KAP1. Oncogene 2008; 27:3054 - 9; http://dx.doi.org/10.1038/sj.onc.1210952; PMID: 18037959
- Kamitani S, Ohbayashi N, Ikeda O, Togi S, Muromoto R, Sekine Y, et al. KAP1 regulates type I interferon/STAT1-mediated IRF-1 gene expression. Biochem Biophys Res Commun 2008; 370:366 - 70; http://dx.doi.org/10.1016/j.bbrc.2008.03.104; PMID: 18381204
- McGough JM, Yang D, Huang S, Georgi D, Hewitt SM, Rocken C, et al. DNA methylation represses IFN-gamma-induced and signal transducer and activator of transcription 1-mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol Cancer Res 2008; 6:1841 - 51; PMID: 19074829
- Cui K, Tailor P, Liu H, Chen X, Ozato K, Zhao K. The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming. Mol Cell Biol 2004; 24:4476 - 86; http://dx.doi.org/10.1128/MCB.24.10.4476-4486.2004; PMID: 15121865
- Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, et al. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 2005; 19:1662 - 7; http://dx.doi.org/10.1101/gad.1323805; PMID: 15985610
- Boneca IG, Dussurget O, Cabanes D, Nahori MA, Sousa S, Lecuit M, et al. A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci USA 2007; 104:997 - 1002; http://dx.doi.org/10.1073/pnas.0609672104; PMID: 17215377
- Trinchieri G. Type I interferon: friend or foe?. J Exp Med 2010; 207:2053 - 63; http://dx.doi.org/10.1084/jem.20101664; PMID: 20837696
- Zenewicz LA, Shen H. Innate and adaptive immune responses to Listeria monocytogenes: a short overview. Microbes Infect 2007; 9:1208 - 15; http://dx.doi.org/10.1016/j.micinf.2007.05.008; PMID: 17719259
- Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol 2008; 181:2092 - 102; PMID: 18641348
- Al Moussawi K, Ghigo E, Kalinke U, Alexopoulou L, Mege JL, Desnues B. Type I interferon induction is detrimental during infection with the Whipple's disease bacterium, Tropheryma whipplei. PLoS Pathog 2010; 6:e1000722; http://dx.doi.org/10.1371/journal.ppat.1000722; PMID: 20090833
- Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol 2010; 84:5670 - 7; http://dx.doi.org/10.1128/JVI.00272-10; PMID: 20335250
- Rayamajhi M, Humann J, Kearney S, Hill KK, Lenz LL. Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence 2010; 1:418 - 22; http://dx.doi.org/10.4161/viru.1.5.12787; PMID: 21178482
- Stockinger S, Decker T. Novel functions of type I interferons revealed by infection studies with Listeria monocytogenes. Immunobiology 2008; 213:889 - 97; http://dx.doi.org/10.1016/j.imbio.2008.07.020; PMID: 18926303
- Cheriyath V, Leaman DW, Borden EC. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J Interferon Cytokine Res 2011; 31:173 - 81; http://dx.doi.org/10.1089/jir.2010.0105; PMID: 20939681
- Mirpuri J, Brazil JC, Berardinelli AJ, Nasr TR, Cooper K, Schnoor M, et al. Commensal Escherichia coli reduces epithelial apoptosis through IFN-alphaA-mediated induction of guanylate binding protein-1 in human and murine models of developing intestine. J Immunol 2010; 184:7186 - 95; http://dx.doi.org/10.4049/jimmunol.0903116; PMID: 20483731
- Potu H, Sgorbissa A, Brancolini C. Identification of USP18 as an important regulator of the susceptibility to IFN-alpha and drug-induced apoptosis. Cancer Res 2010; 70:655 - 65; http://dx.doi.org/10.1158/0008-5472.CAN-09-1942; PMID: 20068173
- Stawowczyk M, Van Scoy S, Kumar KP, Reich NC. The interferon stimulated gene 54 promotes apoptosis. J Biol Chem 2011; 286:7257 - 66; http://dx.doi.org/10.1074/jbc.M110.207068; PMID: 21190939