Abstract
The emergence of Candida strains carrying FKS1 hotspot mutations associated with resistance to echinocandins is cause for concern. However, to assess the potential of such strains to spread within the community and cause lethal infection, the impact of FKS1 mutations on Candida fitness must be determined. We present evidence that C. albicans fks1 mutations carry significant fitness and virulence costs, which are associated with the production of a thickened, chitin-rich cell wall, impaired filamentation and induction of a dampened inflammatory response. If these phenotypic changes remain stable, they can serve as a basis for rational design of strategies to control the spread of echinocandin resistance.
The increasing prevalence of resistance to antimicrobial agents among important human pathogens is rightly regarded as a public health problem of pandemic proportions. The last decade has seen a dramatic rise in the incidence of infections caused by multidrug resistant enterobacteriaceae, methicillin-resistant Staphylococcus aureus, drug resistant human immunodeficiency virus and multidrug or extensively drug-resistant Mycobacterium tuberculosis. The emergence and dissemination of antimicrobial drug resistance threatens to severely limit the availability of therapeutic options for common infections, in effect heralding a post-antibiotic era. However, from an evolutionary stand point, mutations that confer resistance to antimicrobials may impair essential biologic functions or place increased energy demands on the organism, resulting in decreased microbial fitness (Andersson and Hughes, Nat Rev Microbiol 2010). Thus, while microbial strains carrying these mutations possess a survival advantage over wild-type strains while exposed to an antimicrobial drug, in the absence of drug exposure the resistant mutant may be out-competed by drug-susceptible strains. Understanding the interplay between microbial resistance, fitness and virulence, can help make sense of the epidemiology of drug resistance determinants and apply rational measures to limit their spread.
Antimicrobial drugs target essential cellular functions such as cell-wall synthesis, DNA supercoiling, RNA transcription and protein synthesis. The microbial molecules involved in these processes are typically evolutionarily conserved, and hence, mutations that change their affinity to antimicrobial drugs may also affect their function and, as a result, alter microbial fitness. In other cases, upregulation of transmembrane efflux pumps may extrude antimicrobial drugs but also microbial metabolites, thus placing a metabolic burden on drug resistant organisms (Ding et al., J Bacteriol 2008). However, the correlation between resistance and fitness is not as straight forward as might be expected. In fact, the acquisition of a drug resistance determinant by a microorganism may result in a decrease, increase, or no change in fitness (Andersson and Hughes, Nat Rev Microbiol 2010). Moreover, changes in fitness may not remain stable over time; a point mutation that impairs fitness may, over successive generations, be followed by adaptive mutations that compensate for the initial fitness cost, and might eventually result in fitness greater than that of the parental wild-type strain (Cowen et al., Proc Natl Acad Sci USA 2002).
These considerations hold true for pathogenic fungi, and may be of particular importance in this case because, unlike bacteria, fungi do not readily acquire resistance determinants by way of horizontal gene transfer. Thus, the relative fitness of strains with de novo resistance mutations is the principal determinant of the epidemiological success of these strains. Candida species are the most frequent nosocomial fungal pathogens, and the fourth most frequent cause of bloodstream infection in general hospital populations in the US (Wisplinghoff et al., Clin Infect Dis 2004). Invasive candidiasis is associated with high rates of mortality, disability and attendant healthcare costs (Zaoutis et al., Clin Infect Dis 2005). Echinocandins are the newest class of antifungal agents approved for the treatment of candidiasis, and are currently the recommended frontline treatment for invasive candidiasis in critically ill patients (Pappas et al., Clin Infect Dis 2009). Although resistance to these agents is still uncommon, it is increasingly encountered; moreover, the clinical susceptibility breakpoints for echinocandins have recently been revised, resulting in the inclusion of a greater proportion of clinical isolates in the resistant category (Pfaller et al., Drug Resist Updat 2011). Importantly, clinical resistance to echinocandins arises from a limited number of hotspot mutations in the highly conserved FKS genes that encode the 1,3-β-D-glucan synthase (GS) complex. Such mutations have already been shown to alter GS enzyme kinetics; specifically, the maximal catalytic velocity is significantly reduced in FKS mutants (Garcia-Effron et al., Antimicrob Agents Chemother 2009), suggesting that a fitness cost is likely. In recent work (Ben-Ami et al. J Infect Dis 2011), we assessed the effect of FKS1 hotspot mutations on the fitness and virulence of Candida albicans. To that end, we studied laboratory-derived and clinical C. albicans fks1 mutants and their clonal FKS1 wild-type homologs in vitro and in two phylogenetically disparate model host systems, Toll-deficient Drosophila melanogaster and immunocompetent BALB/c mice.
To assess the relative fitness of fks1 mutants and parental wild-types, both strains were allowed to grow together in mixed broth culture for one standard daily growth cycle, after which the relative change in the size of each strain population was determined. We found that even for strain pairs with identical growth rates in single culture, the wild-type population increased by > 600% whereas the fks1 mutant population decreased to ~2% of its original size, indicating attenuated fitness of the echinocandin-resistant strain. We extended this observation to in vivo fitness testing using a mixed infection model in which BALB/c mice were co-infected with uniquely tagged homologous FKS1 mutant and wild-type C. albicans strains. Again, only the wild type strain could be isolated from mouse kidneys 4 d after infection, confirming that the attenuated fitness of the fks1 mutant was important during invasive infection. In terms of virulence, all homozygous fks1 mutants displayed reduced lethality relative to the parental wild-types when injected into Toll-deficient fruit flies. Similar results were obtained in a murine model of hematogenous disseminated candidiasis, which more closely recapitulates invasive candidiasis in humans. In this model, fks1 mutants were associated with significantly higher survival rates and lower fungal burdens in kidneys and spleens compared with the parental wild-type strain.
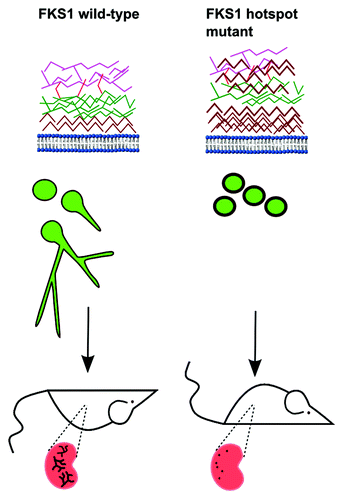
Some clues for the mechanisms underlying these observations may be gleaned from the striking phenotypic changes associated with fks1 mutations in C. albicans. Fks1 mutants had significantly increased cell wall thickness, which was attributed to increased cell wall chitin content (). Synthesis of chitin-rich cell walls is a well characterized end result of activation of cell wall salvage pathways, including the high osmolarity glycerol mitogen activated protein kinase, protein kinase C and Ca2+/calcineurin pathways (Munro et al., Mol Microbiol 2007; Walker et al., PLoS Pathog 2008). Furthermore, fks1 mutants exhibited attenuated growth rates and impaired filamentation capacities (). Morphotypic switching from the yeast (blastoconidia) to the hyphal form plays an important role in C. albicans virulence, as hyphae represent the tissue invasive morphotype, and Candida mutants that are locked in the yeast phase are generally avirulent (Lo et al. Cell 1997; Saville et al. Eukaryot Cell 2003). Interestingly, impaired growth rate and filamentation were most apparent in fks1 mutants with the highest cellular chitin contents. Moreover, the cellular chitin content of fks1 mutants strongly correlated with their reduced virulence in animal model hosts, suggesting that changes in both fitness and virulence in fks1 mutants are linked to the production of a rigid chitinous cell wall, which may be less amenable to remodeling and filamentation. In addition, fks1 mutants elicited a weaker Dectin-1-dependent proinflammatory response than the parental wild-type strain when co-cultured with RAW264.7 macrophages, and this finding too correlated with the cell wall chitin content.
Taken together, these findings indicate that for fks1 mutants, impaired GS enzyme kinetics cause a compensatory increase in cell wall chitin content, which in turn leads to reduced Candida fitness by reducing cell wall plasticity and adaptability. The attenuated virulence of these mutants may be related both to their impaired ability to produce tissue invasive hyphae, and the dampened inflammatory response they produce. Recent work has shown that ultrapure chitin from Candida cell walls blocks the production of proinflammatory cytokines by peripheral blood mononuclear cells in a Dectin-1 dependent manner (Mora-Montes et al. Infect Immun 2011). Thus, over-production of chitin may have abrogated the over-exuberant inflammatory response that is characteristic of disseminated candidiasis.
In interpreting our findings, certain caveats should be kept in mind. First, our analysis was limited to C. albicans, and different associations may exist in other Candida species. Recent analysis of C. glabrata isolates with FKS1 and FKS2 hotspot mutations found more subtle alterations in fitness, which were detected in immunocompetent but not in neutropenic BALB/c mice (Zhao et al., ICAAC 2011). Of note, although C. glabrata is an important emerging human pathogen it is unable to form hyphae, indicating that filamentation is not required for virulence in that species. In another recent study, hotspot mutation in the Aspergillus fumigatus FKS1 gene was associated with attenuated fitness in immunosuppressed mice, and echinocandins maintained modest in vivo activity against the mutant despite in vitro resistance (Lewis et al., J Antimicrob Chemother 2011). Second, our findings may not account for compensatory mutations and chromosomal changes that can occur over time in drug resistant mutants. For example, exposure of C. albicans to fluconazole has been linked with the development of aneuploidy and increased fitness both in the presence and absence of drug (Selmecki et al., PLoS Genet 2009). However, the consistency of our results across a large number of laboratory and clinical isolates suggests that, at least for C. albicans, the attenuated fitness phenotype is stable. Lastly, as mucosal colonization and biofilm formation play important roles in the epidemiology and persistence of Candida species, the interaction of FKS1 mutations with these characteristics is of interest, and has not been addressed here. Further study in this area is ongoing.
Interestingly, our findings are consistent with recent clinical observations. Specifically, analysis of clinical studies of caspofungin for esophageal or invasive candidiasis showed that, paradoxically, favorable treatment outcomes were associated with a caspofungin minimal inhibitory concentration (MIC) > 2 µg/mL (Kartsonis et al., Antimicrob Agents Chemother 2005). It is also notable that C. parapsilosis, a species characterized by relatively high echinocandin MICs as a result of a naturally occurring proline-to-alanine substitution in Fks1p (Garcia-Effron et al., Antimicrob Agents Chemother 2008), displays low GS maximal catalytic velocity and high cell wall chitin content, and is associated with the lowest mortality rate of all Candida species (Horn et al., Clin Infect Dis 2009). These results suggest that in vitro susceptibility testing may fail to predict therapeutic response when resistance is associated with a significant fitness cost. The implications are that, to guide therapeutic decisions, we may need to move beyond traditional in vitro susceptibility testing to more sophisticated analyses that integrate microbial fitness and host factors.
In summary, we have shown that for C. albicans, fks1 mutations that confer resistance to echinocandins are associated with significant fitness and virulence costs. Fitness cost can serve as a basis for strategies to contain the extent of drug resistance in a microbial population by limiting the use of antimicrobial drugs. Thus, restricting echinocandin use should allow susceptible strains to replace less fit fks1 mutants, limiting the potential of echinocandin-resistant strains to spread within hospitals and the community (Andersson et al., Nat Rev Microbiol 2010). Further, fks1 mutants are expected to be associated with a milder clinical course than echinocandin-susceptible strains. Noting the aforementioned caveats however, such predictions should be viewed with appropriate caution. Microbial fitness is a highly dynamic process that is shaped by the forces of evolution. Only the accumulation of epidemiological and clinical data over time will allow us to judge the true impact of fitness on the epidemiology and clinical characteristics of C. albicans fks1 mutants.
Figure 1. Phenotypic changes associated with FKS1 hotspot mutations in Candida albicans. C. albicans FKS1 hotspot mutations are associated with significant increases in cell-wall thickness attributed to increased cell-wall chitin content, impaired growth rate and filamentation capacity. Cell wall components are shown in brown (chitin), green (β-1,3-glucan), red (β-1,6-glucan) and purple (mannan). Production of thick chitinous cell walls likely results from upregulation of chitin synthesis as a result of activation of cell wall salvage pathways. FKS1 mutants exhibit attenuated fitness in vitro, and reduced lethality in immunocompetent BALB/c mice. Excised kidneys show attenuated fungal burdens and the absence of hyphal forms in tissue. It is hypothesized that in fks1 mutants cell-wall chitin blocks Dectin-1 signaling (Mora-Montes et al., Infect Immun 2011), thereby dampening the inflammatory response to β-glucans.