

Abstract
In dialysis-related amyloidosis (DRA), misfolding of β2-microglobulin (β2m) leads to amyloid fibril deposition mainly in the skeletal joints seriously affecting their functionality. The identification and characterization of small-molecules that bind β2m and possibly inhibit its aggregation remain unexplored. In the present study, a ligand-based virtual screening approach and molecular dynamics (MD) simulations were employed to explore potent small-molecule inhibitors against β2m aggregation. The compounds were screened from various small-molecule databases by applying ligand-based virtual screening with rifamycin SV (RSV) as a reference compound. The molecular docking analysis was performed to filter out lead compounds with a higher binding affinity than RSV from a library of ∼800 compounds. Three compounds, ChEBI68321 (C1), ChEMBL360190 (C2) and ZINC3091144 (C3), displaying excellent binding free energies of –51.29, –36.51 and –34.36 kcal/mol, respectively, with β2m were subjected to MD simulations to get insights into the binding locations, key interactions and structural stability of the β2m-ligand complexes. The hydrogen bond analysis highlight higher structural stability and reduced flexibility of the loop regions of β2m in presence of C1, C2 and C3. The integrated computational approach employed in the present study identify promising lead compounds against β2m aggregation in DRA.
| Abbreviations | ||
| β2m | = | β2-microglobulin |
| 3D | = | three dimensional |
| AD | = | Alzheimer’s disease |
| ADT | = | AutoDock Tools |
| DRA | = | Dialysis-related amyloidosis |
| DSSP | = | dictionary of secondary structure of proteins |
| FEL | = | free energy landscape |
| GROMACS | = | GROningen MAchine for Chemical Simulations |
| LGA | = | Lamarckian Genetic Algorithm |
| LINCS | = | LINear Constraint Solver |
| MC | = | main chain |
| MD | = | molecular dynamics |
| MHC–I | = | major histocompatibility complex class I |
| MM–PBSA | = | molecular mechanics Poisson–Boltzmann surface area |
| PME | = | nanometer (nm); particle mesh ewald |
| PCA | = | principal component analysis |
| PDB | = | protein data bank |
| Rg | = | radius–of–gyration |
| RSV | = | rifamycin SV |
| RMSD | = | root–mean–square deviation |
| RMSF | = | root–mean–square fluctuation |
| SC | = | side chain |
| SPC | = | simple point charge |
| SASA | = | Solvent accessible surface area |
| VMD | = | visual molecular dynamics |
Communicated by Ramaswamy H. Sarma
Graphical abstract
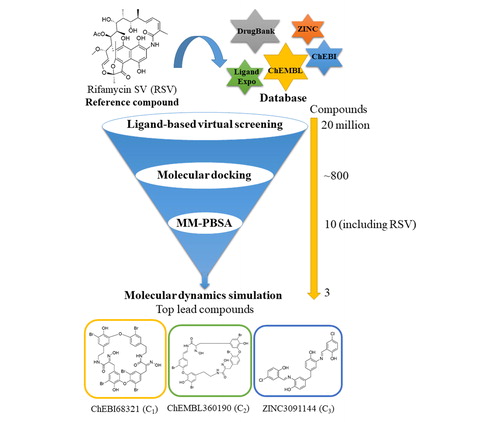
A ligand-based virtual screening approach and molecular dynamics (MD) simulations were employed to explore potent small-molecule inhibitors against β2–microglobulin (β2m) aggregation. The compounds were screened from various small-molecule databases by applying ligand-based virtual screening with rifamycin SV (RSV) as a reference compound. The molecular docking analysis was performed to filter out lead compounds with a higher binding affinity than RSV from a library of ∼800 compounds. Three compounds, ChEBI68321 (C1), ChEMBL360190 (C2) and ZINC3091144 (C3), displaying excellent binding free energies of –51.29, –36.51 and –34.36 kcal/mol, respectively, with β2m were subjected to MD simulations to get insights into the binding locations, key interactions and structural stability of the β2m-ligand complexes. The integrated computational approach employed in the present study identify promising lead compounds against β2m aggregation in DRA.
Acknowledgements
The authors acknowledge C-DAC, Pune for providing the C-DAC's supercomputing resources (PARAM Yuva-II) for the computational facilities. The authors acknowledge School of Chemistry & Biochemistry, Thapar Institute of Engineering & Technology, Patiala, Punjab and Department of Chemistry, Sri Guru Granth Sahib World University, Fatehgarh Sahib, Punjab, India for providing the research facilities.
Disclosure statement
No potential conflict of interest was reported by the authors.