
Figures & data
Figure 1. Risdiplam mechanism of action. The most common form, called 5q-SMA, is caused by a homozygous deletion or loss-of-function mutations in the SMN1 gene on locus 5q13 of chromosome 5, which codes for the homonymous SMN protein A paralogous gene, SMN2, produces functional SMN protein, but at low and insufficient levels due to naturally occurring alternative splicing of its exon 7 that leads to atruncated transcript. The levels of SMN produced from SMN2 can partially compensate for the loss of SMN1. Risdiplam is a small molecule, SMN2 pre-mRNA splicing modifier that promotes the inclusion of exon 7 and production of a full-length SMN2 mRNA, which can then compensate for the loss of SMN1. The working model resulting from studies on risdiplam-like SMN2 splicing modifiers is that the compound binds on two sites within the exon 7 of the SMN2 transcript, namely exonic splicing enhancer 2 (ESE2) and 5′ splice site (5’ss). Binding to the 5′ss enhances the binding of the U1 snRNA. The interaction with the ESE2 is believed to lead to dislocation of the hnRNP G allowing the binding of the U1 snRNP complex. These changes ultimately lead to the inclusion of exon 7 and the production of a full-length SMN2 mRNA. Abbreviations: SMA: spinal muscular atrophy, SMN: survival of motor neuron, ESE2: exonic splicing enhancer 2, 5’ss: 5’ splice site.
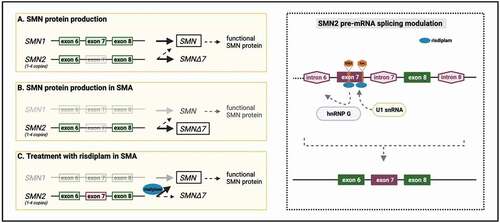
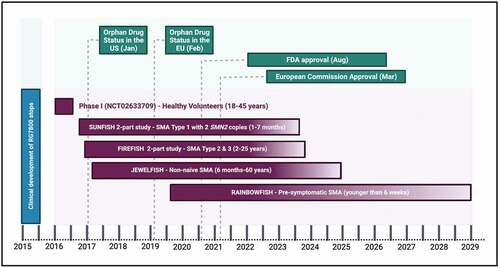
Figure 2. Timeline of risdiplam clinical development and key milestones of approval.
Box 1. Drug summary
Table 1. Summary of clinical trials assessing risdiplam. Abbreviations: PK: pharmacokinetics, PD: pharmacodynamics, SMA: spinal muscular atrophy, SMN2: survival of motor neuron 2, OLE: open-label extension, BSID-III: Bayley Scale for Infant Development-III