Figures & data
Figure 1. Structural and sequence overview of the HU superfamily. (A-D) Cartoon renderings of HU superfamily members. (A) Integration host factor (IHF) α and β in complex with DNA (PDB: 1IHF). IHFα and IHFβ are represented as ribbons and colored green and blue, respectively. DNA is shown as a surface trace in gray. (B) IHFα (PDB: 1IHF_A). (C) HU-HIG clade homodimer from Bacteroides vulgatus with a C-terminal Ig-like domain fusion (PDB: 4FMR). Coloring as in (A) above. The region corresponding to the Ig-like domain is shown as a superimposed ribbon with surface representation colored in gray. (D) HU domain from chain A of Bacteroides vulgatus HU homolog (PDB: 4FMR_A). The domains are colored and labeled as in (B), with additional secondary structure elements colored white. (E) Multiple sequence alignment of the HU superfamily. Secondary structure provided in top line, with elements labeled to correspond with (B). Positions shown to interact with DNA are denoted by asterisks. Sequences are labeled to left with NCBI accession number and organism abbreviation separated by rightmost underscore; HU family/clade names are given to the right. Negative numbers at left indicate extension of predicted protein start sites in GenBank. The alignment is colored as follows: h, hydrophobic and yellow; l, aliphatic and yellow; s, small and green; p, polar and blue; u, tiny and green. Organism abbreviations: Esili, Ectocarpus siliculosus; Pfalc, Plasmodium falciparum; Otaur, Ostreococcus tauri; Ehuxl, Emiliania huxleyi; Gsulp, Galdieria sulphuraria; Plunu, Pyrocystis lunula; Kvene, Karlodinium veneficum; Acart, Amphidinium carterae; Ptetr, Paramecium tetraurelia; Tcruz, Trypanosoma cruzi; Ssalm, Spironucleus salmonicida; Ngrub, Naegleria gruberi; Mcomm, Micromonas commoda; Hsapi, Homo sapiens; Nvect, Nematostella vectensis; Aplat, Anas platyrhynchos; Ggall, Gallus gallus; Pging, Porphyromonas gingivalis; Ctrac, Chlamydia trachomatis; Cbact, Cytophagaceae bacterium; Bsp, Bacteroides sp, Dsp, Dysgonomonas sp; Bfrag, Bacteroides fragilis; Gbact, Gallionellales bacterium; Tdent, Treponema denticola; Fsp, Flavobacterium sp; Prumi, Prevotella ruminicola; Btimo, Bacteroides timonensis; Bfine, Bacteroides finegoldii; Pgula, Porphyromonas gulae; Psp, Prevotella sp.
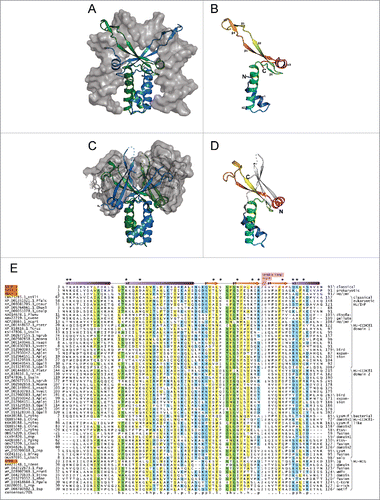
Figure 2. Phylogenetic relationships and genome associations in HU-CCDC81 and HU-HIG families. (A) Phylogenetic tree depicting higher-level relationships between HU families/clades described in this study. Branches are collapsed at levels containing clearly-delineated monophyletic groups, labeled to the right. Nodes with greater than 65% bootstrap support are marked with yellow circle. Representative conserved domain architectures and gene neighborhoods in a given clade provided to the right. (For complete list see Supplemental Material). Phyletic patterns of a given architecture/neighborhood are found provided to the right in green lettering. Phylogeny abbreviations: b, bacteroidetes; C, Chlamydia; P, Porphyromonas; s, spirochaetes; β, β-proteobacterial; γ, γ-proteobacteria; δ, δ-proteobacteria; v, verrucomicrobia; a, animals; k, kinetoplastids; api, apicomplexa; diplo, diplomonads; N, Naegleria; cili, ciliates; chloro, chlorophytes; stram, stramenopiles; SAR, stramenopile-alveolate-rhizarian group; Phy, Phytopthora; o, oomycetes; G, Guillardia. (B) Phylogenetic tree depicting the multiple paralogs identified in avian expansion of HU-CCDC81 domains. Monophyletic clades, as determined by phyletic distribution conservation patterns, are collapsed and then labeled and colored according to evolutionary depth. Nodes with greater than 70% bootstrap support are marked with yellow circle. Potential lineage-specific expansions within a clade are labeled with total number of non-redundant protein copies and phyletic patterns. (C) Phylogenetic tree depicting rampant LSEs, gene loss, and incomplete lineage-sorting in HU-HIG family based on a set of all HU-HIG sequences retrieved from the 10 bacteroidetes genomes, listed in key to the right, with the highest number of identifiable HU-HIG sequences. Branch coloring in tree corresponds to genome name colors in key. Domain architectures typical of sequences in clustered branches ring the tree, see (A) for explanation of architecture depictions. Complete trees provided in Newick format in Supplemental Material.
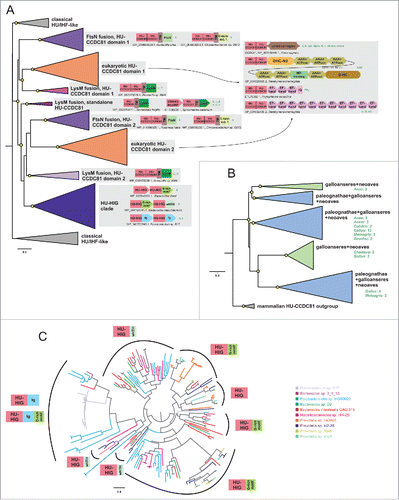
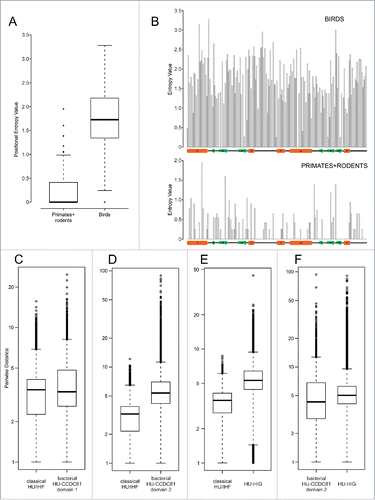
Figure 3. Positional entropy and sequence diversity comparisons. (A) Positional entropy comparison between Gallus and Meleagris HU-CCDC81 domains (galliform birds) and primate and rodent HU-CCDC81 domains. Entropy values calculated as described in Materials and Methods. (B) Entropy values from (A) plotted along linear sequence of HU-CCDC81 domain, secondary structure provided below and labeled in concordance with . (C-F) Sequence diversity plots comparing pairwise sequence evolutionary distances (see Materials and Methods) within representatives of labeled HU families, y-axes set to log scale. Differences in boxplots (A, C-F) are significant (p < 2.2e-16) by Wilcoxon rank sum test.