Figures & data
Figure 1. RefSeq LOC100288798 is a ubiquitously expressed, inefficiently processed lncRNA (A) Overview of the genomic locus. UCSC Genome Browser screenshot – from top to bottom: CpG island annotation, RefSeq Genes annotation, GENCODE v19 annotation, UCSC Genes annotation, MiTranscriptome lncRNA transcripts,Citation2 Cabili et al lincRNA transcriptsCitation17.(B) LOC100288798 is a ubiquitously expressed lncRNA. Heat map shows expression level of SLC38A2, SLC38A4 and LOC100288798 (marked as “lncRNA” throughout the figure) in multiple tissues and cell types. Letters in brackets after the name of each sample indicate the source and the type of RNA-seq (see Table S1A for details of abbreviations). Expression levels of SLC38A4 and LOC100288798 were calculated as average RPKMs of RefSeq isoforms (SLC38A2 – 1 isoform: NM_018976, SLC38A4 – 2 isoforms: NM_018018 and NM_001143824, LOC100288798 – 5 isoforms: NR_125377, NR_125378, NR_125379, NR_125380, and NR_125381), values are displayed inside each cell. Heat map color legend is displayed on the left. (C) LOC100288798 lncRNA is variably spliced in different tissues. Heat map shows splicing efficiency (Methods) of LOC100288798 and 2 protein-coding genes TPB, SLC38A2 (well-spliced ubiquitously expressed protein coding gene controls) in publicly available total RNA-seq data (Table S1A). Calculated splicing efficiency is displayed inside each cell. Heat map color legend is displayed on the left. (D) Visual inspection of ENCODE HeLa RNA-seq of various cell and RNA fractions suggests that LOC100288798 is an inefficiently processed lncRNA. From top to bottom: Chromosome position; RefSeq annotation; ENCODE HeLa RNA-seq sequencing data. RNA-seq data is displayed using the public ENCODE RNA-seq (CSHL) hub in the UCSC browser (only Replicate 2 from 2 replicates available at ENCODE RNA-seq (CSHL) hub is displayed). From top to bottom: PolyA+ RNA-seq of the whole cell Reverse and Forward strand show absence of SLC38A4 expression from the reverse strand and visible expression from the forward strand corresponding to LOC100288798. Dashed orange lines indicate chromosome positions of RefSeq annotated exons of LOC100288798. Comparison of signal intensities between polyA+ and polyA- indicates LOC100288798 is inefficiently spliced as it appears more abundant in polyA- fraction. Cytoplasm RNA-seq indicates that only spliced and polyadenylated LOC100288798 transcripts can be exported to the cytoplasm (compare peaks in polyA+ and no peaks in polyA-). Nuclear RNA-seq indicates nuclear enrichment of LOC100288798 unspliced form (compare nucleus polyA- to cytoplasm polyA-). RNA-seq tracks are displayed with the default ENCODE RNA-seq (CSHL) hub scale (range - from 0 to 100). (E) PolyA+ enrichment. Bar plot shows PolyA+ enrichment (calculated as the ratio between RPKM in PolyA+ and PolyA- RNA fractions) of the 4 indicated genes in HeLa cells (ENCODE RNA-seq data). RPKMs and consequently PolyA+ enrichment were calculated for spliced isoforms (RPKM over exons, blue bars) and unspliced isoforms (RPKM over whole gene body, purple bars) of the 4 genes. PolyA+ enrichment is a relative value, therefore we indicated the absolute RPKM values of spliced and unspliced isoforms in PolyA- fraction below each respective bar. (F) Nuclear enrichment. Bar plot shows nuclear enrichment (calculated as the ratio between RPKM in nuclear and cytoplasmic fractions) of the 4 indicated genes in HeLa cells (ENCODE RNA-seq data). RPKMs and consequently nuclear enrichment were calculated for spliced isoforms (RPKM over exons, blue bars) and unspliced isoforms (RPKM over whole gene body, purple bars) of the 4 genes in PolyA+ (darker bars) and PolyA- (lighter bars) fractions. Nuclear enrichment is a relative value, therefore we indicated the absolute RPKM values in cytoplasmic fraction below each respective bar.
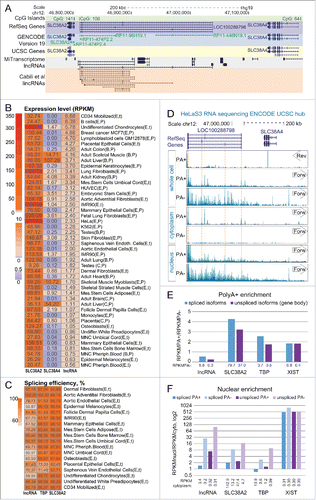
Figure 2. LOC100288798 exon structure assembly from various tissues extends its annotation to over 500kb overlapping SLC38A4.UCSC Genome Browser screen shot of the studied locus (chr12:46,772,500-47,422,500). From top to bottom: Chromosome position and the scale; RefSeq gene annotation (all annotated isoforms are displayed), spliced human ESTs (12/35 ESTs displayed), transcriptome assembly of the locus obtained in this study (Results, Methods). Note that only selected transcripts are shown (11/167 de novo isoforms of LOC100288798 and 4/43 de novo isoforms of SLC38A4), and that both EST and transcriptome assembly data reveal extension of LOC100288798 to over 500kb in length. RNA-seq tracks from ENCODE/CSHL UCSC hub with the titles containing cell type name, RNA-seq type and transcriptional orientation are displayed below. Only total whole cell RNA-seq is displayed. Bottom: normalized RNA-seq signal from wild type human haploid KBM7 cell lines (merged data from 2 wild type clones sequenced in this study, Methods). For all RNA-seq tracks: only forward strand (Plus Signal) is displayed.

Table 1. Stop cassette insertions overview.
Figure 3. Gene trap technology allows truncation of SLC38A4-AS lncRNA in human haploid KBM7 cell line (A) Overview of the experimental design: SLC38A4-AS truncation and control cell lines used in the study. Top row: Wild type KBM7 cells underwent the gene trap insertion procedure and single clones were selected and expanded to a monoclonal population. Three independently obtained clones with gene trap cassettes mapping within the gene body of SLC38A4-AS lncRNA were available (see ). Two monoclonal cell lines with independent insertion events that integrated a gene trap cassette 3kb downstream of SLC38A4-AS transcription start site (TSS) were available (3kb1 and 3kb2). Only one monoclonal cell line had a gene trap insertion 100kb downstream of the downstream of SLC38A4-AS TSS. Therefore we prepared biological replicates by performing independent thawing and culturing procedures (100kb1 and 100kb2). Left column: We obtained 3 wild type KBM7 control cell lines, which did not undergo any gene trap insertion procedure, were not monoclonal and were cultured by different people at different times prior to culturing for this analysis (WT1, WT2 and WT3). Middle column: To control for changes during gene trap insertion and selection procedure we obtained 2 KBM7 cell lines that did undergo gene trap insertion within the body of HOTTIP lncRNA and were monoclonally expanded (C1 and C2) (see ). (B) Ploidy of KBM7 cell lines assessed by cell size. Bar plot shows peak cell size measured for 9 cultured KBM7 cell lines (Methods). All the cell lines were thawn and processed in one batch by the same person. Cell size was measured at the first splitting (3 days post-thawing, dark gray bars), second splitting (6 days post-thawing, medium gray bars), and prior to harvesting (8 days post-thawing, light gray bars). (C) Ploidy of KBM7 cell lines assessed by total DNA amount. Bar plot shows total DNA mass isolated from 20 million cells. DNA mass in the plot is normalized to WT1 sample (absolute value for WT1 is 109 μg). (D) Confirmation of successful SLC38A4-AS truncation by RT-qPCR. Top: schematic representation of the locus (drawn to scale). Blue bars show RefSeq annotation of LOC100288798 and SLC38A4 genes. Black bar underneath shows the extended annotation of LOC100288798 (SLC38A4-AS) obtained in this study (). White arrows inside the bars indicate transcriptional orientation of the gene. Below the positions of stop cassette insertions () and RT-qPCR probes are displayed (). Bottom: Expression profiling of SLC38A4-AS in the KBM7 cell lines (described in A). Error bars represent standard deviation from 3 RT-qPCR technical replicates. Bars are ordered from left to right as listed (top to bottom) in the legend on the right. For each RT-qPCR probe the expression level in WT1 is set to 100%.
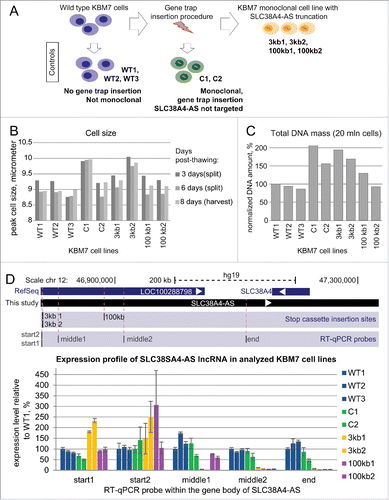
Table 2. RT-qPCR probes for analyzing expression profile of SLC38A4-AS lncRNA.
Figure 4. RNA-seq confirms truncation and continuity of the SLC38A4-AS lncRNA gene. (A) SLC38A4-AS RNA-seq signal of the 8 clones analyzed in D. Top: schematic representation of the locus (as described for D). Bottom: RNA-seq signal, normalized to sample read number, pink dots indicate RNA-seq signal that exceeds the range presented inside the box. Type of the cell line is indicated on the left, name of the cell line is indicated on the right. Vertical dashed red lines indicate position of the 3kb and 100kb stop cassettes. Low density of RNA-seq signal piles indicate low expression and the smallest size corresponds to 1 read. (B) Expression profile of different regions of SLC38A4-AS lncRNA in the RNA-Seq data shown in (A). Bar plots show RPKM of the regions of SLC38A4-AS indicated on the X axis for 4 types of cell lines (as grouped on A). RPKM value for each clone type is averaged from 2 cell lines, error bars show the RPKM values of the 2 samples. Numbers above the bars show the plotted value. Note that this analysis allows the comparison of regions within one cell line but not between cell lines. (C) Expression profile comparison of SLC38A4-AS between analyzed clones. Bar plot shows RPKM of the regions of SLC38A4-AS indicated on the X axis for each cell line type normalized to the value for “Wild type”. Normalized RPKM values are the average of 2 cell lines of each type, indicated by the error bars.
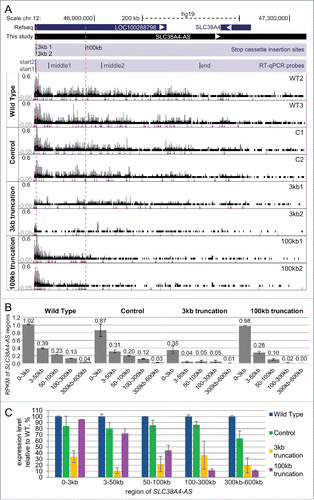
Figure 5. Genome-wide differential expression analysis reveals deregulation of protein-coding genes in trans upon SLC38A4-AS lncRNA truncation (A) Expression level of genes differentially expressed between SLC38A4-AS truncation cell lines and the 4 control cell lines allows unsupervised clustering of the cell lines that resembles the different cell groups. Heat map shows expression level (FPKM, Methods) of genes (name indicated on the right) with significant differential expression (p < 0.01, >3 fold expression change, Methods) between 2 conditions: no SLC38A4-AS truncation (WT2, WT3, C1, C2) and genetic truncation of SLC38A4-AS (3kb1, 3kb2, 100kb1, 100kb2). Expression values are normalized to the mean FPKM among all 8 samples. Mean is set to 1. Names of genes that form the filtered stringent list of deregulated genes (, Methods) are displayed in bold blue font. Heat map color legend is displayed on the right. (B) and (C) Examples of up- and downregulated protein coding genes from the stringent list (). CD9 is markedly upregulated (B) and RORB is markedly downregulated (C) upon truncation of SLC38A4-AS. UCSC Genome Browser screen shots show normalized RNA-seq signal. Top to bottom: Chromosome position, RefSeq gene annotation, RNA-seq signal, normalized to sample read number, from eight sequenced cell lines. Each box shows the same range from 0 to 0.6, only forward strand is shown. Pink dots indicate RNA-seq signal that exceeds the range presented inside the box. Name of cell line is indicated on the left.
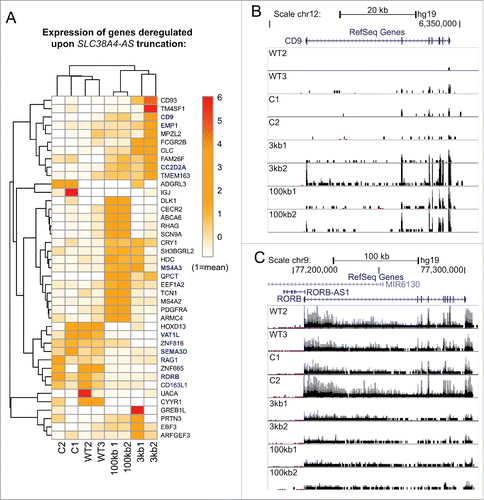
Table 3. Stringent list of genes affected by SLC38A4-AS lncRNA truncation.
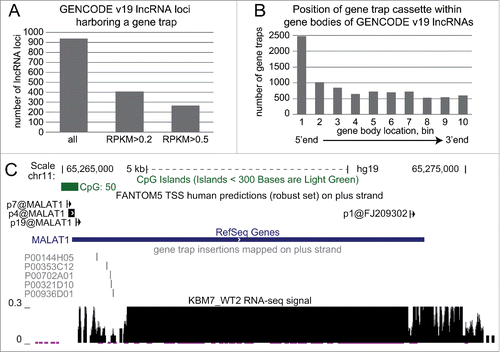
Figure 6. Haploid gene trap collection represents a rich resource for quick functional assessment of hundreds of lncRNAs. (A) Hundreds of GENCODE v19 lncRNAs expressed in KBM7 cell line are targeted by a gene trap insertion. Bar plot shows number of non-overlapping GENCODE v19 lncRNA loci that contain a gene trap cassette in the same transcriptional orientation in KBM7 clones within the “Human Gene Trap Mutant Collection” (left bar, Methods), and the number of these lncRNA loci that are expressed (middle bar, loci that contain lncRNA transcripts expressed with RPKM > 0.2) and well expressed (right bar, loci that contain lncRNA transcripts expressed with RPKM > 0.5) in wild type KBM7 cells. (B) Gene trap cassettes are preferentially inserted at the 5’ end of lncRNAs. Bar plot shows the number of gene trap cassettes inserted into different regions in the gene bodies of GENCODE v19 lncRNA. Numbers correspond to 10 equally sized, non-overlapping regions investigated for each gene. (C) Five genetic truncations of the well-known lncRNA MALAT1 are available within the “Human Gene Trap Mutant Collection." Shown is the UCSC browser screen shot of the MALAT1 gene region. From top to bottom: chromosome scale, CpG island annotation (UCSC track), FANTOM5 TSS predictions (robust set)Citation82 on the plus strand, RefSeq gene annotation, position of gene trap insertion cassettes available (plus strand), normalized RNA-seq signal from WT2 KBM7 cell line showing wild type expression of MALAT1.