
Figures & data
Figure 1. Process for classifying non-coding RNA (ncRNA). First, we gather a diverse dataset in the “data collection” step. Next, in “handcrafted features”, we pull out specific details from the data. Then, in “feature encoding”, we adjust these details to prepare them for the final step. Lastly, in “classification”, we use modern computer models that combine different techniques to classify RNA.
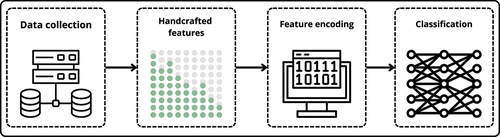
Figure 2. Illustration of the data collection pipeline that uses infernal to extract the desired ncRNA sequences from genomes for training and testing. The ’cmfetch’ function retrieves covariance models from the complete Rfam database, and ’cmsearch’ matches these models against a genome. The output consists of multiple FASTA files, each corresponding to a specific ncRNA class. This method is based on the genomic pipeline in Bonidia et al. [Citation51].
![Figure 2. Illustration of the data collection pipeline that uses infernal to extract the desired ncRNA sequences from genomes for training and testing. The ’cmfetch’ function retrieves covariance models from the complete Rfam database, and ’cmsearch’ matches these models against a genome. The output consists of multiple FASTA files, each corresponding to a specific ncRNA class. This method is based on the genomic pipeline in Bonidia et al. [Citation51].](/cms/asset/625fb576-e4ab-4e29-bbbf-a5a9e50efafd/krnb_a_2329451_f0002_oc.jpg)
Table 1. Number of sequences used for training and testing.
Figure 3. Illustrative depiction of feature encoding techniques for genomic data, focusing on the integration of one-hot encoding, dictionary encoding, and k-mer representation. A hypothetical nucleotide sequence is employed to demonstrate these methods. For one-hot encoding, each nucleotide is mapped to a unique four-dimensional binary vector, while ordinal numbers are used to represent each nucleotide in dictionary encoding. The figure also presents examples of k-mer one-hot and k-mer dictionary encoding, where sequences are divided into subsequences of length ’k’ and encoded using the respective methods.
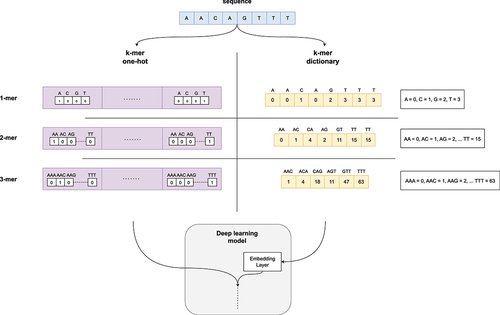
Figure 4. Schematic representation of the proposed ML pipeline for biological sequences classification, integrating CNN and BiLSTM networks. The pipeline initiates with the data loading phase and proceeds with sequence encoding using methods such as k-mer one-hot and k-mer dictionary. Following resource extraction, data is fed into a hybrid deep-learning model comprised of convolutional and LSTM blocks. To accommodate k-mer dictionary encoding, the data is passed through an embedding layer, while convolutional layers extract spatial patterns and LSTMs capture temporal dependencies. Dropout layers are optionally included to prevent overfitting. After processing through LSTMs, data is flattened and directed to fully connected layers for final output generation. The pipeline can integrate multiple encodings and concatenating handcrafted features, allowing for efficient classification of biological sequences. The model parameters and structure were set based on prior empirical work and related literature.
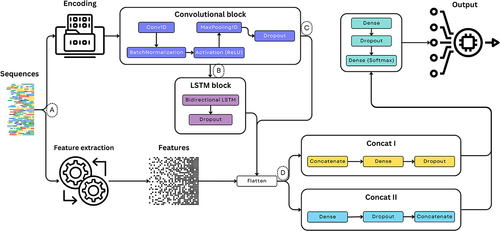
Table 2. Weighted precision results obtained with different variants of the CNN architecture. The models are categorized based on the encoding method (k-mer one-hot (enc I), k-mer dictionary (enc II), and their combination (enc I + enc II)), the number of convolution blocks, and the concatenation approach (direct concatenation (concat I) and concatenation with a dense layer for each branch (concat II)). Results are compared for 1-mer, 2-mer, and 3-mer configurations. Results surpassing those of traditional classifiers are highlighted in bold.
Figure 5. Comparative performance analysis of our proposed architectures against traditional machine learning classification models (SVM and XGBoost), considering the best configurations obtained. (a) Performance comparison of the algorithms using the weighted average of metrics such as precision, recall, and F1-score. The results show consistency throughout the metrics and better performance for CNN with external features. (b) Performance comparison by class using the precision obtained with the algorithms. Even though classes such as rRNA seem to be classified effortlessly, there are clear improvements in precision in classes such as sRNA and cis-regulatory elements.
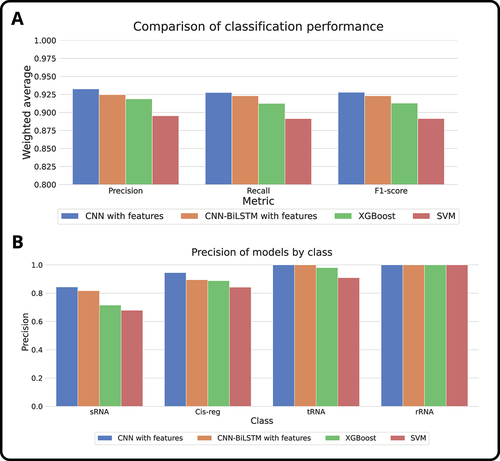
Table 3. Weighted precision results obtained with different variants of the CNN-BiLSTM architecture. The models are categorized based on the encoding method (k-mer one-hot (enc I), k-mer dictionary (enc II), and their combination (enc I + enc II)), the number of convolution blocks, and the concatenation approach (direct concatenation (concat I) and concatenation with a dense layer for each branch (concat II)). Results are compared for 1-mer, 2-mer, and 3-mer configurations. Results surpassing those of traditional classifiers are highlighted in bold.