Figures & data
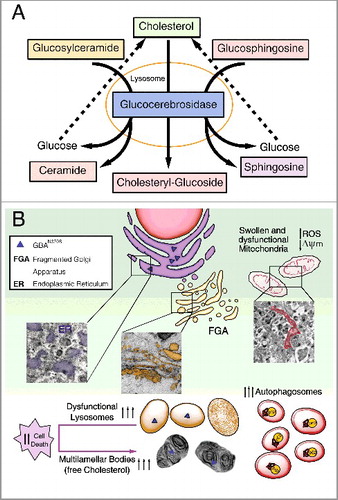
Figure 1. Diagram summarizing the primary altered features caused by GBAN370S. (A) In physiological conditions GBA hydrolyzes its primary substrate glucocerebroside into glucose and ceramide. An alternate substrate, glucosylsphingosine, is also degraded into glucose and sphingosine. GBA also acts as a glycosyltransferase, catalyzing the transfer of glucose from glucocerebroside to cholesterol and leading to the formation of cholesteryl-glucoside. (B) Normal lysosomal function is required for the autophagic clearance of defective cellular organelles and misfolded proteins. The N370S mutation results in GBA loss of function in the lysosomes (caused by its retention in the ER) and accumulation of its substrates. This defect leads to stress, enlargement and disorganization of the ER, and Golgi apparatus fragmentation (FGA), along with subsequent defects in autophagy shown as an autophagosome accumulation. Accordingly, SQSTM1 accumulates due to dysfunctional lysosomes likely caused by cholesterol accumulation that promotes MLB formation. These alterations hamper the removal of damaged mitochondria, inducing ROS production. All this together renders these cells vulnerable to stress-induced apoptosis.