Figures & data
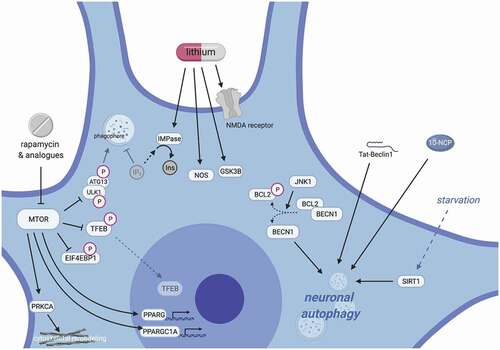
Figure 1. Dysfunction of autophagy-related proteins impairs proteostasis and leads to neurotoxicity in ALS. (A) Under normal conditions, SQSTM1 serves as a receptor protein in selective autophagy and binds both LC3-II and polyubiquitinated proteins, thereby targeting ubiquitinated substrates to phagophores (left); Mutations in SQSTM1 abrogate SQSTM1’s binding activities (right top) or result in the aggregation of SQSTM1 into ubiquitin-positive inclusions (right bottom). (B) The C9orf72 protein participates in several autophagy-related complexes, including the autophagy induction complex (ULK1-RAB1A) that promotes autophagosome biogenesis, the RAB7-RAB11 complex (RAB complex) that regulates endosome maturation, and the C9orf72-SMCR8-WDR41 (CSW) complex that regulates lysosomal dynamics and autophagic flux (left). Disease-associated C9orf72 mutations reduce C9orf72 protein levels (right), while dipeptide repeat proteins generated from the C9orf72 expansion localize to SQSTM1- and ubiquitin-positive inclusions (right). (C) In normal mitophagy, TBK1 binds and phosphorylates OPTN, enhancing its affinity for polyubiquitinated mitochondria and LC3-II (left). TBK1 and OPTN mutations perturb these functions and compromise efficient mitophagy, leading to failed mitochondrial clearance and dysfunctional mitochondria.
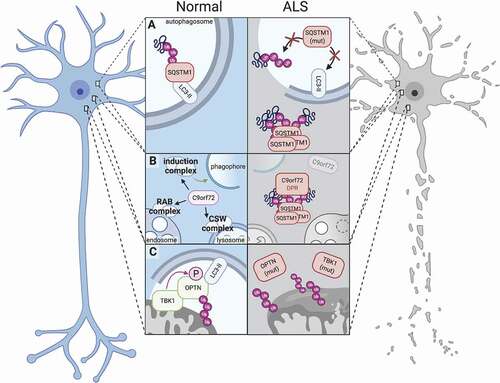
Table 1. Role of starvation-induced MTOR inhibition in various cell types of the nervous system
Figure 2. Distinct factors regulate autophagy among different cell types of the nervous system. In each of the cells which comprise the central and peripheral nervous systems, autophagy is differentially regulated by cell type-specific effectors. In neurons (top left), modulation of SPHK1 by phenoxazine compounds such as 10-NCP, but not starvation, potently induces autophagy. In contrast, nutrient deprivation is sufficient to promote SPHK1 signaling and autophagy induction in astrocytes (top center). In the axonal compartment of neurons, KIF1A and PINK1-PRKN are critical for facilitating local autophagic activity (left, middle). MicroRNAs such as MIR101 and MIR195 suppress oligodendrocytic and Schwann cell autophagy, respectively (right). Schwann cells also clear myelin debris through myelinophagy, a unique form of selective autophagy that is dependent on FIG4 (bottom). In muscle, specific transcription factors can exert activating (FOXO3) or suppressive (PPARGC1A, RUNX1) effects on autophagy, and phosphatases such as MTM1 and MTMR14 inhibit autophagy by recycling phosphoinositides needed for autophagy induction (bottom left).
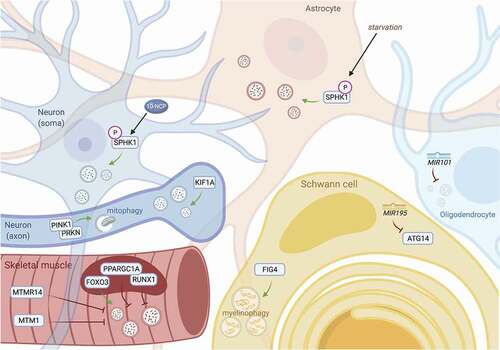
Figure 3. Mechanisms of neuronal autophagy and their impact for therapeutic design in ALS. Pharmacodynamic considerations limit the use of currently available drugs for modulating autophagy. Inhibitors of MTOR inhibitors (rapalogs) only weakly activate autophagy in neurons, and their wide-ranging effects target myriad cellular pathways, including growth signaling, translation, stress responses, transcriptional regulation, and cytoskeletal remodeling (left). Such pleiotropy leads to well-documented and multi-systemic toxicities, especially with long-term use; however, newer rapalogs may enable more specific targeting and selective autophagy modulation. Similarly, lithium has numerous multi-target effects, including depletion of IP3 through IMPase, activating nitric oxide synthase, inhibiting GSK3B, stimulating NMDA receptors and enhancing glutamatergic tone, among many others (middle). This results in a narrow therapeutic index for lithium, potentially explaining its apparent lack of neuroprotective effects in human trials for neurodegenerative disease to date. To reduce off-target effects, recent efforts have focused on MTOR-independent strategies for stimulating autophagy, including modulation of SIRT1, phenoxazine compounds such as 10-NCP, or synthetic peptides such as Tat-Beclin 1 (right).