Figures & data
Figure 1. Challenges for single-stage allele replacement strategies using CRISPR. (A) HDR using a ssODN as repair template is shown. Because ssODNs are limited to ∼200bp, the sgRNA target site (shown in green) must be in close proximity to the sequence to be edited, as the ssODN must span the repair site and have homology to both sides. Unless the introduced change disrupts the target site, the edited locus may be re-cleaved by Cas9, potentially leading to error-prone NHEJ repair. (B) HDR using a dsDNA plasmid as the repair template is shown. If the genomic target site sequences (shown in blue and purple) are present in the donor plasmid, Cas9 cleavage may lead to cutting and subsequent degradation of the donor plasmid; successful repair events will also be vulnerable to cleavage (C) A second mutation at the PAM site (NGG → N*G) may be introduced to prevent re-cleavage of the edited sequence, but this additional mutation(s) may have undesired phenotypic effects. (D) Mutating the PAM sites (NGG → N*G) prevents unwanted cleavage of dsDNA donor or repaired genomic sequence, but involves the addition of extra mutations known as “scars” that may affect phenotypes. Abbreviations used in the figure are defined as follows: NGG = protospacer-adjacent motif; CCN = reverse complement (opposite strand) PAM sequence; N*G or C*N = mutated protospacer-adjacent motif; ssODN = single-stranded oligodeoxynucleotide donor; HDR = Homology-directed repair; HA = Homology arm. Scissor symbols represent target sites expected to be cleaved.
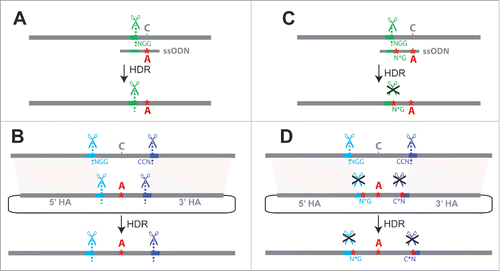
Figure 2. Schematic of marker-assisted, 2-stage allele swap within the D. americana tan transgene in D. melanogaster. In stage 1, the 3′ end of the first exon (black rectangle) and a portion of the first intron of D. americana tan were excised by cleavage at the t5 (antisense direction) and t3 sgRNA target sites shown in brown. In the donor plasmid used to repair this region, pGEM-WingGFP-tan, the PAM sites (highlighted in yellow) and 3 PAM-proximal nucleotides at each target site contain sequence from the native target site, but the remaining 17 nucleotides of each sgRNA target sequence have been edited to differ from the D. americana tan sequence. These edited sequences serve as new, unique CRISPR target sites for reporter excision, and are labeled as t5re and t3re (“re” for “reporter excision”). These t5re and t3re target sites are not recognized by the sgRNA-Cas9 complexes targeting sites t5 and t3, thus preventing cleavage of pGEM-WingGFP-tan or the HDR product. When the wing-GFP transformation marker was incorporated into the genome, so were these unique t5re and t3re target sites, which contain restriction sites that double as multiple cloning regions. The donor plasmid used for stage 2, pGEM-tan-edits, contained the region of the D. americana tan sequence amplified with primers shown as arrows labeled A and B, which was cloned into the pGEM T-Easy vector. Changes in the length of 2 homopolymer runs used to confirm genome modification are represented by red asterisks. sgRNAs targeting the t5re and t3re sites flanking the reporter gene were used to remove it, with the D. americana tan sequence restored from pGEM-tan-edits via HDR. Locations of PCR primers used to test flies that lost wing-GFP expression following stage 2 of the allele swap are shown with arrows labeled X and Y. Precise HDR was confirmed by Sanger sequencing the amplicon produced by these primers. For primer sequences and details about screening PCRs, see Table S1 in the supplement. Sanger sequencing chromatograms for all edited sites are shown in Figure S1.
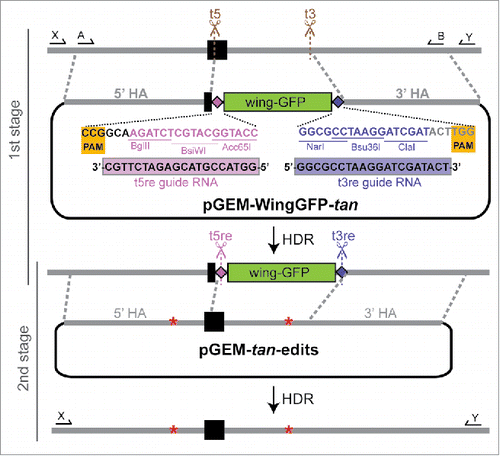
Figure 3. Representative pigmentation and fluorescence phenotypes of flies at each stage of the tan allele swap process. (A) Dorsal pigmentation of adult flies is shown for the melDA tan strain prior to editing (left), the melDA tan strain in which the targeted region of tan has been replaced with the wing-GFP marker (middle), and the melDA tan strain after the wing-GFP marker was replaced with the edited tan sequence (right). The darker pigmentation seen in flies on the left and right is caused by a functional D. americana tan transgene. When this transgene is disrupted (middle), pigmentation is visibly lighter on the dorsal head, thorax, and abdomen. Double-headed black arrows indicate areas in the thorax and abdomen where the change in pigmentation was most readily apparent. (B) GFP fluorescence in late-stage pupae is shown for the melDA tan strain prior to editing (left), the melDA tan strain in which the targeted region of tan has been replaced with the wing-GFP marker (middle), and the melDA tan strain after the wing-GFP marker was replaced with the edited tan sequence (right). The GFP fluorescence in eyes of all 3 flies results from the 3XP3-GFP reporter gene included in the D. americana tan transgene. GFP expression in the developing wings (indicated with arrows) is visible in flies after the first stage of the 2-step allele swap procedure (middle) and lost following the second stage (right). All pupae shown were deemed to be at the same developmental stage based on visible features of wing development, expression of 3XP3-GFP, and lack of pigmentation on the developing wing.
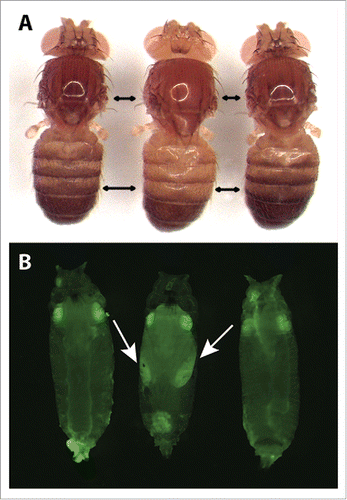
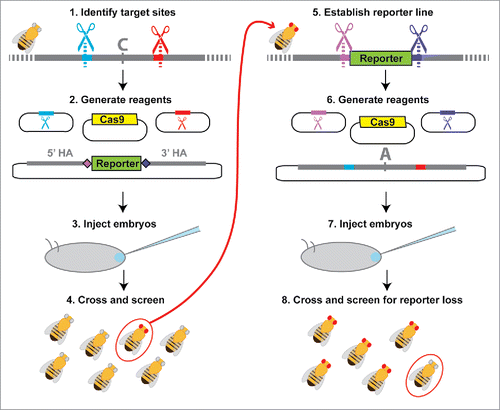
Figure 4. Workflow for 2-stage marker assisted allele swap. (1) Target sites flanking the area to be edited are identified (red and blue) using online tools to identify optimal target sites and search for potential off-target cleavage sites.Citation22,39 (2) Sequences from the selected target sites are then cloned into sgRNA expression plasmids or used to generate in vitro transcribed sgRNAs. Homology arms flanking the region of interest (recommended length ∼1kb) are cloned into the reporter donor plasmid, which contains unique CRISPR target sites (light and dark purple) in place of the genomic target sites (red and blue). (3) Embryos are injected with the donor plasmid, sgRNAs (expression plasmids or in vitro transcribed RNAs) and a source of Cas9 (expression plasmid, mRNA, or protein) unless any of these components is produced by a transgene already present in the host. (4) Adult flies that develop from the injected embryos are collected as virgins and then crossed back to the parental line. F1 progeny emerging from these crosses are screened for the selectable marker, with flies positive for the selectable marker allowed to produce F2 progeny before extracting their DNA for molecular screening. (5) Individuals with the correct reporter gene insertion are made homozygous and the population is expanded for embryo collections. (6) sgRNAs with sequences matching the unique CRISPR target sites introduced with the reporter gene (light and dark purple) as well as a plasmid containing the original CRISPR target sites and the edited version of the original sequence are prepared. (7) Flies carrying the reporter at the locus of interest are injected for the second allele swap step. (8) Because the selectable marker is dominant, adult flies developing from injected embryos must be crossed back to either the original parental line from step (1) or to a balancer line (if available) for screening. Progeny from this cross that do not show expression of the selectable marker are then crossed and analyzed with molecular tests to determine whether they contain the desired editing events.