Figures & data
Figure 1. Inhibition and activation of TNFR1. TNFR1 is strongly activated by its natural, trimeric ligand TNF. The bivalent IgG ATROSAB was shown to exert a dominant antagonistic activity in the presence of TNF, yet on its own exerts a marginal TNFR1 activation in a narrow dose range. Monovalent formats like the Fab or the newly developed Fv-Fc1k are effective antagonists of TNF mediated TNFR1 activation and lack intrinsic agonistic activity. In addition, the Fv-Fc1k comprises an Fc proportion, providing a prolonged serum half-life, comparable to an IgG.
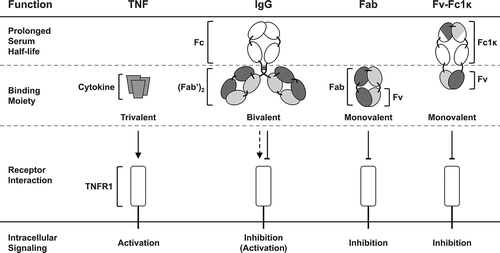
Figure 2. Generation of a heterodimerizing Fc with FcRn-interacting regions. A novel heterodimerizing Fc was generated by replacing the IgG1 CH3 domains with interspersed constant Ig domains, based on the per se heterodimerizing constant Ig domains CH1 and CLκ, complemented with elements of IgG1 CH3 domains, that are responsible for the interaction with the FcRn. The newly generated heterodimerizing Fc was designated Fc-one/kappa (Fc1k).
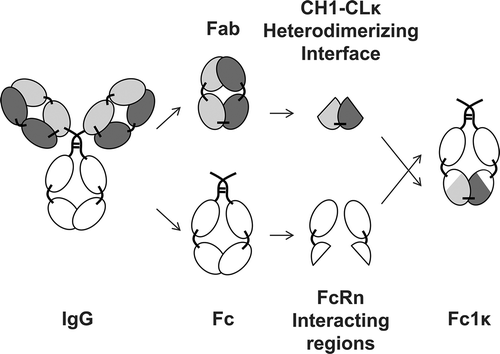
Figure 3. Proof of concept of Fc1k using asymmetric scFv-Fc1k fusion proteins. An scFv fragment was fused to the N-terminus of the hinge region of an Fc chain, containing either the newly generated CH31 or CH3κ domains, the heterodimerizing CH3knobs or CH3holes domains, or the wild type CH3 domains. Proteins were analyzed by SDS-PAGE (NuPAGETM 4–12% Bis-TRIS Midi Gel). Descriptions: M (Marker), 1: (scFv connected to Hinge-CH2-CH31); 2: (scFv connected to Hinge-CH2-CH3κ); 3: (scFv connected to Hinge-CH2-CH3knobs); 4: (scFv connected to Hinge-CH2-CH3holes); 5: (scFv connected to Hinge-CH2-CH3wt). Staining: Coomassie Brilliant Blue, de-staining: Water.
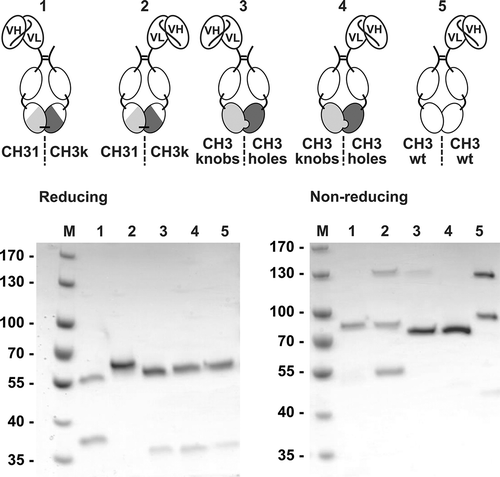
Figure 4. Generation and optimization of Atrosimab. Two versions of a novel Fv-Fc1k fusion protein were generated, (a) composed of the chains VH-CH2-CH31 (VH1C)/VL-CH2-CH3κ (VLkC) or (b) composed of VL-CH2-CH31 (VL1C)/VH-CH2-CH3κ (VHkC). Both molecules were compared in SDS-PAGE (c and d, NuPAGETM 4–12% Bis-TRIS Midi Gel) under reducing (R) and non-reducing conditions (NR) and SEC (e and f, Phenomenex Yarra SEC-2000, 300 × 7.8 mm, flow rate of 0.5 ml/min, mobile phase Na2HPO4/NaH2PO4).
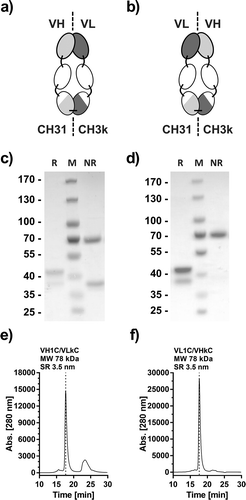
Figure 5. Biochemical characterization of Atrosimab. Atrosimab was produced by Catalent (Catalent Pharma Solutions, Somerset, Ewing, NJ, US) from a CHO cell pool after stable lentiviral transduction and purified by protein A chromatography and subsequent gel filtration (performed at the University of Stuttgart). Characterization was performed by analytical SEC (a, TSKgel SuperSW mAb HR, Flow rate 0.5 ml/min, mobile phase Na2HPO4/NaH2PO4) and SDS-PAGE (b), NuPAGETM 4–12% Bis-TRIS Midi Gel under reducing (R) and non-reducing conditions (NR). M: Marker. (c) The melting temperature was determined by dynamic light scattering and visual interpretation of the obtained results. Plasma stability was analyzed after incubation in human plasma for the indicated time points followed by the determination of the EC50 values of residual binding protein by ELISA (D).
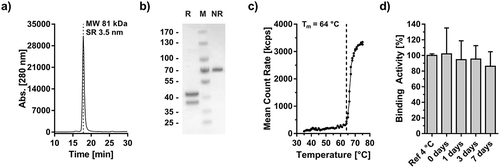
Figure 6. Antigen, Fc receptor and Complement binding of Atrosimab. Binding of Atrosimab to human TNFR1-Fc was analyzed in ELISA (a, n = 3, mean ± SD) and QCM (b). Five concentrations between 128 nM and 4 nM (1:2 dilution steps) were used to generate the kinetic data in b. A one-to-one binding algorithm was employed for fitting. C) Binding of human FcγRIa, IIb and IIIa and the complement protein C1q to immobilized Atrosimab was analyzed by ELISA. Rituximab (wild-type Fc part) and ATROSAB (silent Fc) were used as controls (n = 2, mean ± range).
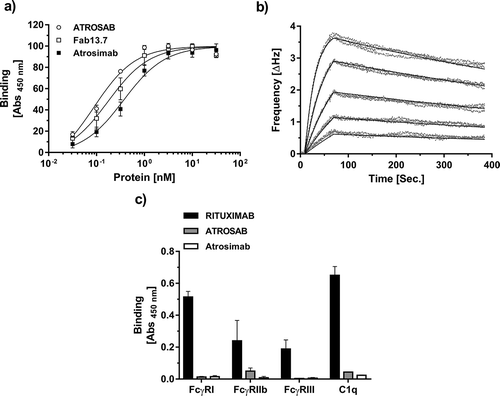
Figure 7. Antagonistic bioactivity of Atrosimab and lack of agonism. The inherent lack of agonistic activity of Atrosimab in terms of TNFR1 activation was demonstrated in three individual assays. a) IL-6 release from HeLa cells, b) IL-8 release from HT1080 cells and c) cell death induction assay using Kym-1 cells. The inhibitory potential of Atrosimab was shown in an IL-6 release assay using HeLa cells (d), in an IL-8 release assay using HT1080 cells (e) and in a cell death induction assay using Kym-1 cells (f), which were performed in the presence of a constant concentration of 0.1 nM TNF (d and e) or 0.01 nM TNF (f). ATROSAB (marginal activity) and TNF (strong activity) alone served as control molecules for the activation of TNFR1, Fab 13.7 served as negative control (a, b and c). Fab 13.7 and ATROSAB served as controls for the inhibition of TNF-induced TNFR1 activation (d, e and f). All graphs represent the mean of three individual experiments, error bars indicate SD.
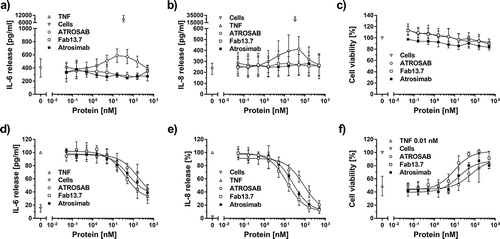
Table 1. Bioactivity of Atrosimab.
Figure 8. Complete lack of agonistic bioactivity of Atrosimab in the presence of anti-human IgG antibodies. The activation of TNFR1 on the surface of HT1080 cells by Atrosimab in the presence of a constant concentration (ca. 15.8 nM) of three different anti-human IgG serum preparations (a, b and c) was determined by the detection of IL-8 release into the culture supernatant. Unstimulated cells and 33 nM TNF were used as controls. The agonistic effect of potentially crosslinking antibodies was compared to Fab 13.7 and ATROSAB. All experiments show Mean ± SD of three individual experiments.
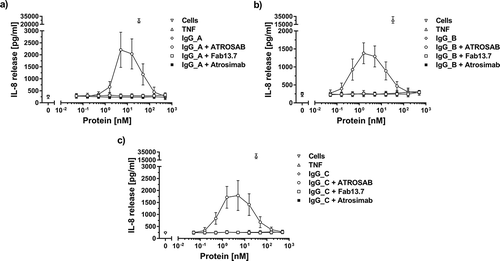
Table 2. Pharmacokinetic analysis of Atrosimab.
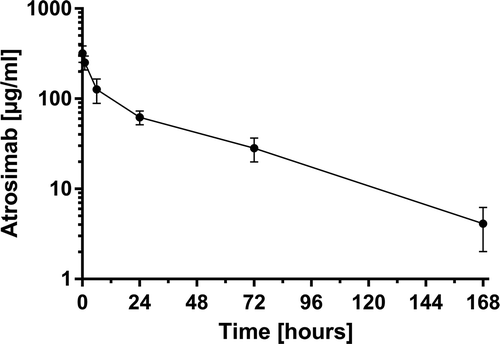
Figure 9. Pharmacokinetic analysis of Atrosimab. Four hundred micrograms of Atrosimab were injected into C57BL/6J knock-in mice, carrying the gene of the human TNFR1 extracellular domain connected to the mouse transmembrane and intracellular domains instead of the wild-type mouse gene. Remaining intact protein in the serum was determined by ELISA for binding to TNFR1 at the indicated time points. Shown are the mean ± SD of five mice.