
Figures & data
Figure 1. Enrichment of canine scFv phage display library after panning against cPD-1 antigen. Recombinant cPD-1 was adsorbed to a microtiter plate overnight at 4°C. Wells were washed and blocked with 2% milk in PBS (MPBS) for 1 hour at 37°C. Initial unpanned library (P0) and libraries of phage obtained after each round of selection (P1 through P4) were added to coated plates and incubated for 1 hour at 37°C. Plates were washed with PBS supplemented with 0.1% Tween (PBST) and bound phage was detected using a 1:5000 dilution of HRP-conjugated anti-M13 mAb in MPBS. Bound phage were detected with ABTS. OD was read at 405 nm after 30 min using a Molecular Devices SpectraMax 340 spectrophotometer.
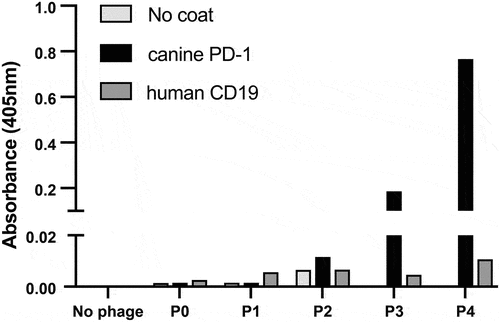
Figure 2. Binding of randomly selected scFv-expressing monoclonal phage clones from panning rounds 3 and 4 to cPD-1. ELISA plates were coated with recombinant cPD-1 and human CD19 (left) or canine IL-13 Rα2 (right) as controls. Polyclonal phage from the fourth round of panning against cPD-1 served as positive control phage on both plates. Polyclonal phage from the third round of panning against human CD19 (left graph) and fourth round of panning against canine IL-13Ra2 (right graph) (both sets of phage from unrelated projects) served as positive phage controls. Each monoclonal phage was screened once for binding to cPD-1.
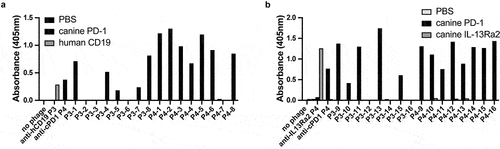
Figure 3. The ability of unique soluble scFv clones to bind to cPD-1 by ELISA. 19 unique soluble scF-vs were tested for binding to cPD-1 by ELISA. Increasing amounts of biotinylated cPD-1 were added to streptavidin coated ELISA plate wells and incubated for 1 hour. 0.25 μg/ml soluble HA-tagged scFvs were added to the wells and bound scFvs were detected using an AP-conjugated anti-HA antibody. An irrelevant soluble HA-tagged scFv against MERS (Middle Eastern Respiratory Syndrome) virus from an unrelated project was used as a negative control. A. unique clones identified in the initial survey. B. additional unique clones identified via high throughput screen. Evaluation of each unique soluble clone against increasing amounts of biotinylated cPD-1 was performed once.
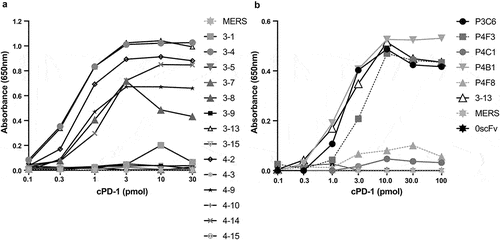
Figure 4. PD-1:PD-L1 inhibition assay. A. Schematic of experimental setup. Soluble biotinylated cPD-1 was incubated with increasing concentrations of cPD-L1-Fc either alone or in the presence of a selected soluble anti-cPD-1 scFv or an irrelevant soluble scFv against Middle Eastern Respiratory Syndrome (MERS) virus. Incubation mixtures were added to ELISA plates pre-coated with streptavidin to capture cPD-1:cPD-L1-Fc complexes. Bound complexes were detected using an anti-Fc-AP conjugate and AP colorimetric substrate. B. soluble anti-cPD-1 scFvs show varying degrees of inhibition of cPD-1 binding to cPD-L1 as revealed by a loss of detectable signal.
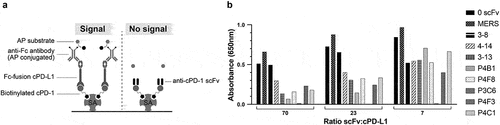
Figure 5. Soluble canine anti-cPD-1 scFv binding to cell surface expressed cPD-1. The human erythroleukemic cell line K562 was gene edited to eliminate the FcγRII (CD32) (KTδ32). Retroviral transduction of edited cells to express cPD-1 was performed, and cPD-1 positive target cells were selected in puromycin. Soluble, purified, HA-tagged scFvs were incubated with target cells, and bound scFvs were detected using a fluorescent anti-HA antibody. The soluble scFv against MERS and scFv 3–7 (negative binders to cPD-1 by ELISA), were used as negative controls.
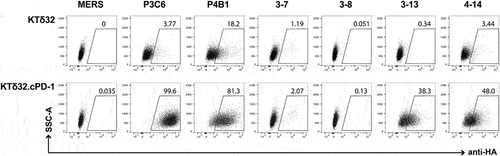
Figure 6. A. Primary structures of lead canine anti-cPD-1 antibodies. A. (top) heavy chain alignments of P4B1, P3C6 and its variants along with canine germline gene IGHV3-38*01. (bottom) light chain alignments of P4B1 and P3C6. Framework and CDR regions use IMGT nomenclature. B. Evaluation of full-length IgGD (P3C6mut3.1 and mut3.2) canine anti-cPD-1 binding to cell surface-expressed cPD-1 by flow cytometry. Canine IgGD-HA tagged clones against cPD-1 or the irrelevant MERS antigen (negative control) were used to stain KTδ32 and KTδ32.cPD-1 cells. Bound IgGD molecules were detected using an anti-HA antibody. Plots are gated on live, 7AAD- cells.
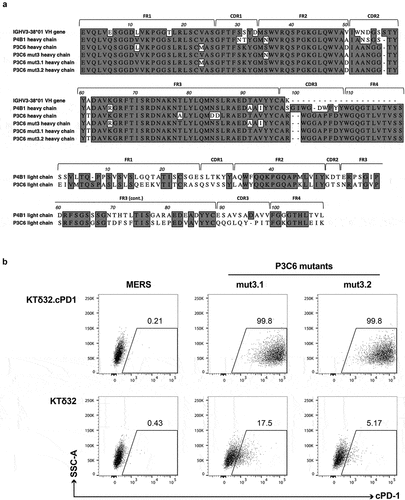
Figure 7. Full-length IgGD P3C6mut3.1 reverses the effects of PD-L1 inhibition on canine CAR-T cells. A. K562 cells were engineered to express canine CD20 (K562-cCD20) and canine PD-L1 (K562-cCD20-cPD-L1) and cell surface expression was confirmed by flow cytometry. B. Canine CD20 CAR-T cells from one dog were labeled with cell trace violet (CTV) and co-cultured at an E:T ratio of 1:1 with either K562-cCD20 or K562-cCD20-cPD-L1 in the presence of either anti-MERS or P3C6mut3.1 IgGD. After 72 hours of culture, proliferation of CD8+ CAR-T cells was assessed by flow cytometry. Plots are gated on live CD5+ CD8+ CAR+ cells. C. Canine CD20 CAR-T cells from one dog were co-cultured at an E:T ratio of 1:1 with the same target cells as in A. Expression of CD107b was determined after 4 hours of co-culture. Plots are gated on live CD5+ CD8+ CAR+ cells.
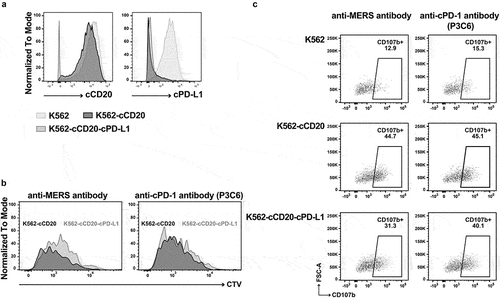
Figure 8. Pharmacokinetics of full-length IgGD/B P3C6mut3.1 in healthy dogs. Healthy dogs were administered P3C6mut3.1 IgGD/B intravenously at either 2 mg/kg (n = 2) or 10 mg/kg (n = 2) on Day 0 and Day 21. Blood samples were taken at the indicated time points and analyzed for the presence of P3C6mut3.1 IgGD/B in the serum using a custom Meso Scale Discovery immunoassay. A. Serum pharmacokinetics of P3C6mut3.1 IgGD/B with data represented as mean ± SD. B. Dose-normalized concentration vs. time profiles for P3C6mut3.1 IgGD/B.
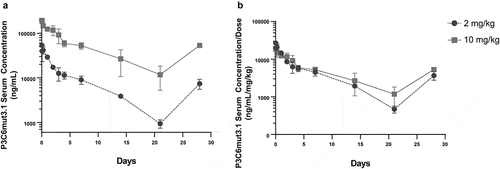
Table 1. A non-compartmental analysis of full-length P3C6mut3.1 IgGD/B PK parameters following the first dose. Cmax: maximum observed serum concentration, AUClast: area under the serum concentration vs. time curve from time 0 to 21 days, AUCinf: area under the serum concentration vs. time curve extrapolated to time infinity, t1/2: terminal log-linear half-life, CL: clearance, Vss: volume of distribution.
{kind=link}
{kind=link}