Figures & data
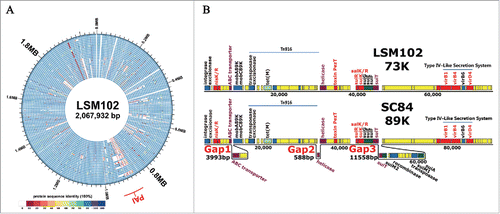
Figure 1. (A) Geographic distribution and sequence types of human S. suis isolates from 2006-2016 in this study. One symbol represents an isolate. Closed symbols denote isolates collected in this study, and open symbols denote isolates reported by literatures or MLST database. (B) The phylogeny of 26 completely sequenced S. suis strains. The Maximum likelihood tree was estimated by the SNPs of a core genome contained 885 genes. Bootstrapping was conducted using 500 replicates. The SC84 strain of S. suis was used as a reference. The label of each branch orderly showed the sequence type, the country and the host from which the S. suis strain was obtained. Then it marks whether or not the strain harbors the 89K PAI, salKR or nisKR (+ : the complete 89K PAI is included, salKR or nisKR; − : the complete 89K PAI is absent, salKR or nisKR; T : partial of 89K PAI associated region is detected).
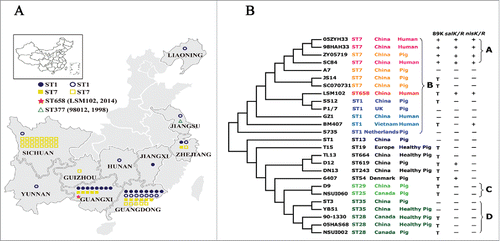
Figure 2. (A) Genomic comparison of 26 S. suis genomes. The circular diagrams showed the variations of LSM102 compared with the other completely sequenced S. suis strains. From outside to the inside, the order of the genomes was: LSM102, SS12, P1/7, ZY05719, SC84, 98HAH33, 05ZYH33, A7, SC070731, JS14, GZ1, BM407, S735, ST1, TL13, D12, DN13, T15, 6407, NSUI060, D9, NSUI002, 05HAS68, 90-1330, YB51, ST3. The different colors stand for the percentage of protein sequence identity. (B), Gene organization of 73 kb pathogenicity island (73K PAI) of LSM102. Virulence-related factors including SalKR, NisKR and Type IV–Like Secretion System (VirD4, VirB1, VirB4) that have been reported to be involved in full virulence of STSS-causing Chinese S. suis were included in 73K PAI of LSM102. Compared with 89K, 3 fragment losses/deletions occur in 73K PAI of LSM102, which were designated as Gap1, Gap2, and Gap3 respectively.