Figures & data

Figure 1. Life cycle of RV. (1) The invading RV TLPs attach to receptors [sialoglycans and Histo-blood group antigens (HBGAs)] and co-receptors [integrins and heat shock cognate protein 70 (hsc70)] assembled in lipid rafts on host cellular plasma membrane. (2) RV TLPs are endocytosed and trafficked into the cells. (3) TLPs are uncoated to form DLPs which are released from endosomes into the host cellular cytoplasm. (4) Within DLPs, viral RNAs are transcribed to yield capped, (+)ssRNAs. (5) RV (+)ssRNAs are translated on host cellular ribosomes to synthesize viral proteins (NSPs and VPs) which are necessary for evading innate antiviral immune response and (6) nucleation of RV-specific inclusion bodies called viroplasms. Host cellular lipid droplets (LDs) act as scaffolds for viroplasm formation. (7) Inside the maturing viroplasms, viral genome replication takes place within the VP2-encaged viral cores through the VP2-driven polymerase activity of VP1. Progeny cores acquire the VP6 layer and form progeny DLPs which may either amplify the replication cycle by producing secondary transcripts or (8) enter into the morphogenetic assembly pathway. Acquisition of the outer capsid occurs by a budding step through the ER-derived cellular membrane where VP6 on DLPs docks on NSP4 on ER-derived membrane. Subsequently, NSP4 is stripped and VP7-VP4 layer is assembled. Alternatively, acquisition of VP4 spikes may occur on VP7-surrounded virions within ER-Golgi intermediate compartment (ERGIC)/plasma membrane lipid raft domains (9a) before non-lytic virion release. (9b) RV progenies may also exit through lytic mechanisms
![Figure 1. Life cycle of RV. (1) The invading RV TLPs attach to receptors [sialoglycans and Histo-blood group antigens (HBGAs)] and co-receptors [integrins and heat shock cognate protein 70 (hsc70)] assembled in lipid rafts on host cellular plasma membrane. (2) RV TLPs are endocytosed and trafficked into the cells. (3) TLPs are uncoated to form DLPs which are released from endosomes into the host cellular cytoplasm. (4) Within DLPs, viral RNAs are transcribed to yield capped, (+)ssRNAs. (5) RV (+)ssRNAs are translated on host cellular ribosomes to synthesize viral proteins (NSPs and VPs) which are necessary for evading innate antiviral immune response and (6) nucleation of RV-specific inclusion bodies called viroplasms. Host cellular lipid droplets (LDs) act as scaffolds for viroplasm formation. (7) Inside the maturing viroplasms, viral genome replication takes place within the VP2-encaged viral cores through the VP2-driven polymerase activity of VP1. Progeny cores acquire the VP6 layer and form progeny DLPs which may either amplify the replication cycle by producing secondary transcripts or (8) enter into the morphogenetic assembly pathway. Acquisition of the outer capsid occurs by a budding step through the ER-derived cellular membrane where VP6 on DLPs docks on NSP4 on ER-derived membrane. Subsequently, NSP4 is stripped and VP7-VP4 layer is assembled. Alternatively, acquisition of VP4 spikes may occur on VP7-surrounded virions within ER-Golgi intermediate compartment (ERGIC)/plasma membrane lipid raft domains (9a) before non-lytic virion release. (9b) RV progenies may also exit through lytic mechanisms](/cms/asset/d259d955-bde8-41b5-8750-edb31447b7ee/kvir_a_1903198_f0001_oc.jpg)
Figure 2. Entry of RV TLPs into host cells. The entire event of ingress of RV TLPs across the host cell membrane into the cytoplasm is an elaborate process which consists of (1) attachment and (2) post-attachment interactions between the virions and the host cells, followed by (3) endocytosis, (4) endosomal trafficking and penetration of the virus into the host cytosol in the form of DLPs. (1) Host cell membrane sialylated (SA-containing) and fucosylated (HBGAs) glycans act as receptors for RV TLPs. NA-sensitive RV strains require terminal SA-containing glycans as entry receptors; NA-resistant RV strains utilize sub-terminal SA-containing glycans and HBGAs. The RV spike protein VP4, especially by its trypsin-digested domain VP8, interacts with the cellular receptors. (2) Cellular integrins (α2β1, α4β1, αXβ2, αVβ3) and hsc70 act as post-attachment co-receptors for RV TLPs. A DGE motif of RV-VP5 which is a trypsinized fragment of RV-VP4 is required for interaction with α2β1 integrin. Additional VP5 and VP7 regions are required for interactions with hsc70 and integrins. Cellular receptors and co-receptors for RV entry are spatially assembled in lipid raft microdomains of cell membrane. (3) Virions are subsequently internalized by clathrin-mediated or clathrin-independent endocytosis. The GTPase dynamin, cellular actin microfilaments, several actin-binding proteins (Actn4, Diaph, drebrin), and actin microfilament regulatory components (RhoA, Cdc42) modulate the process of rotaviral entry. Viral tethering activates RhoA-dependent stress fiber formation and TJ disruption downstream of RhoA/ROCK/MLC signaling. (4) Endocytosed virions converge in EEs and may progress on to the LEs for uncoating and penetration (late penetrating RVs) or may be released before that (early penetrating RVs). Therefore, early penetrating RVs are sensitive to EE markers EEA1 and Rab5 but not to LE markers Rab7 and Rab9 whereas late penetrating RVs lose infectivity in absence of Rab7 and Rab9. ESCRT complex responsible for ILV formation is also essential for RV entry process. Late penetrating RV strains require protein transporters CD-M6PRs which deliver cathepsins from TGN to LEs. Other protein transporters such as CI-M6PRs and sortilin also regulate infectivity of late penetrating RV strains. TLPs are uncoated to form DLPs which are released from endosomal compartment into host cytosol. Endosomal acidification and calcium concentration can act as triggers for viral nucleocapsid exit. PI3K and ERK signaling downstream of interaction between cell surface receptor/co-receptors and late penetrating RV strains can activate the V-ATPase proton pump resulting in endosomal acidification and viral uncoating. Several inhibitors acting on different steps of RV entry are shown. For additional details, please refer the text
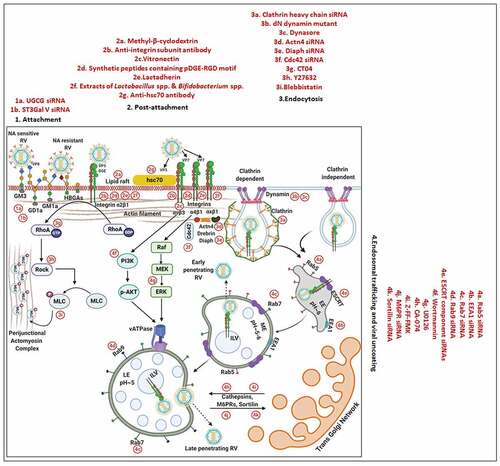
Figure 3. Evasion of host cellular antiviral responses by RV. (a) Evasion of host cellular IFN response. Within RV infected cells, RV RNA species with exposed 5ʹ-phosphate groups or with incomplete 5ʹ-O-methylated “cap” structures act as PAMPs and are recognized by host cellular PRRs RIG1 and MDA5. Subsequent oligomerization of the mitochondrial adaptor MAVS forms a platform for recruitment of TRAFs and two kinase complexes IKK-α/β/γ and TBK1-IKKε leading to the activation of IRF3/7 and NF-κB-dependent transcriptional programme (IFNs, ISGs, cytokines). NF-κB contains two subunits p50 and p65. IKK-α/β/γ phosphorylates NF-κB inhibitory protein IκB at the α subunit resulting in its proteasomal degradation by SCFβ-TrCP (Skp1-Cullin1-F-box containing protein β-TrCP) E3 ubiquitin ligase complex. Being freed of IκB, p50-p65 heterodimeric NF-κB translocates to nucleus and causes transcriptional activation. RV-NSP1 targets many host proteins of this pathway proteasomally (such as MAVS, β-TrCP, IRF3/7, TRAF2) or non-proteasomally (RIG1) and causes their degradation in a RV strain-dependent manner. RV-VP3 has also been shown to target MAVS for proteasomal degradation. Moreover, at least for β-TrCP degradation, a fostering role of hijacked host cellular Cul3-Rbx1 E3 ubiquitin ligase machinery has been implicated. NSP1-β-TrCP interaction also requires CK-II-directed NSP1 phosphorylation reactions. Some RV strains block NF-κB function through sequestration of NF-κB p65 away from nucleus into viroplasmic puncta. The amplification step of IFN response includes binding of IFNs (type I and type II) to cognate receptors (type I IFN receptor IFNAR and type II IFN receptor IFNGR) to activate JAK-STAT signaling. The phosphorylated forms of STAT1 and STAT2 form a complex called ISGF3 by associating with IRF9. This heterotrimeric complex translocates to the nucleus, binds to IFN-stimulated response elements (ISREs) and trans-activates a series of ISGs of cyto-protective and antiviral nature. Homodimeric phosphorylated STAT1 downstream of IFNGR signaling also trans-activates ISGs by nuclear translocation and binding to Gamma interferon activation site (GAS). RV infection curtails IFN amplification pathway by promoting degradation of IRF9, IFN receptors, and by preventing STAT1 phosphorylation as well as STAT1-STAT2 nuclear translocation. RV-NSP1 is responsible for degradation of IRF9 proteasomally and for inhibition of STAT1 phosphorylation. (b) Evasion of host cellular OAS/RNase L pathway by RV. Within virus infected cells, viral dsRNA population can induce oligomerization-dependent activation of the enzyme OAS which further catalyzes the formation of 2ʹ-5ʹAs. Upon interacting with 2ʹ-5ʹAs, RNase L gets activated through dimerization and triggers cleavage of RNA including the viral RNA species. RV evades the deleterious effects of OAS/RNase L pathway by its structural protein VP3. RV-VP3 has intrinsic 2ʹ, 5ʹ-phosphodiesterase (2ʹ-5ʹPDE) motif through which it disintegrates 2ʹ-5ʹA structures, thereby preventing RNase L activation and viral RNA cleavage. (c) Evasion of innate antiviral impacts of host RNAi machinery by RV. Double stranded RV replication intermediates can potentially be subjected to trimming by host cellular DICER resulting in production of virus‐derived small interfering RNAs (viRNAs). Incorporation of viRNAs into the RISC containing the catalytic effector AGO2 can subsequently target viral RNA population. During early hours of RV-SA11 and RV-A5-13 infection, RV-NSP1 interacts with and ubiquitylates AGO2 leading to proteasomal demise of this catalytic effector. In this respect, RV-NSP1 acts as a putative viral‐suppressor‐of‐RNAi (VSR). In absence of AGO2, siRNA/shRNA-guided RNAi and also potential viRNA-directed RNAi are rendered nonfunctional during early hours of RV infection. Clonal overexpression of AGO2 shows anti-RV effects. (d) Attenuation of host cellular anti-oxidant defense system by RV. Nrf2 is the master transcription factor which deals with cellular redox stress by transcribing anti-oxidant and cyto-protective effectors such as HO-1, NQO1, SOD1. Under unstressed condition, Nrf2 is constantly turned over in a ubiquitin-proteasome-dependent way by cellular Keap1-Rbx1-Cul3 machinery. (Left panel) In RV infected cells, reactive oxygen species (ROS) is induced during early hours leading to Keap1 inhibition and Nrf2 upregulation. Elevated Nrf2, further primed by PKC-mediated phosphorylation, translocates to nucleus and trans-activate stress responsive genes which contain Nrf2-binding motif [anti-oxidant response element (ARE)] in their promoter regions. Quenching ROS by NAC has an antagonizing effect on RV infection. (Right panel) During later hours of RV infection, Nrf2 is expelled out of the nucleus, ubiquitylated by a non-canonical E3 ubiquitin ligase (other than the canonical Keap1-Rbx1-Cul3 machinery) and degraded proteasomally. Levels of HO-1, NQO1, and SOD1 were also reduced. Agonists of Nrf2/ARE pathway, such as Keap1 inhibitors CDDO-Me and RA-839, show potent anti-RV effects. (e) Time-dependent regulation of host cellular apoptotic cell death by RV. (Left panel) During early hours of infection, anti-apoptotic pathways are activated and pro-apoptotic pathways are inhibited for ensuring viral replication. The prime most survival pathway includes activation of PI3K-Akt signaling as a result of interaction of RV-NSP1 with PI3K. Interaction of the chaperone Hsp90 with Akt has an agonistic effect on this pathway. Inhibition of survival pathways through targeting PI3K, phospho-Akt and Hsp90 by LY, 294–002, triciribine and 17-AAG, respectively, sensitized RV replication. Inhibition of pro-apoptotic pathways is multifaceted. One of them is the upregulation of the miRNA population hsa-miR-142-5p by RV-NSP5. Elevated hsa-miR-142-5p sensitizes its targets TGFβR II and SMAD3 leading to attenuation of p38MAPK-ERK1/2-JNK-dependent apoptotic signaling in HT29 cell line. Another strategy is the ubiquitylation and proteasomal degradation of p53 during early infection period. RV-NSP1 plays a pivotal role in this regulation. In absence of p53, transcription of p53-dependent apoptotic genes is prevented. Yet another anti-apoptotic modality in early hours of RV infected cells is the prevention of RV-NSP4 translocation to mitochondria. This is enabled by the ubiquitin-proteasome-dependent demise of the mitochondrial chaperonin Hsp60 which facilitates NSP4 mitochondrial import. A phosphorylation event of Hsp60 carried out by the autophosphorylated and activated form of Src kinase (SrcY416) imparts proteasomal sensitivity to Hsp60. Targeting hsa-miR-142-5p by its anti-miR and Src kinase by a small molecule SKI-I exert anti-RV activity. (Right panel) During late phase of infection, apoptotic pathways are activated and/or de-repressed and outweigh the survival pathways. Intrinsic pathway of apoptosis observed in late hours of RV infected cells is partially dependent on Bax. Subsequent release of cytochrome c into cytosol results in apoptosome formation and activation of executioner caspases. Reduced level of hsa-miR-142-5p results in de-repression of TGFβR II-SMAD3-p38MAPK-ERK1/2-JNK-dependent apoptotic signaling in HT29 cell line. Weakened interaction of p53 with RV-NSP1 stabilizes p53 and causes p53-dependent transcription of pro-apoptotic genes (PUMA, Bax. Bak). In absence of SrcY416, Hsp60 is no longer phosphorylated and therefore escorts NSP4 across mitochondria. NSP4 also positively regulates apoptotic mitochondrial fragmentation by promoting Cdk1-dependent phosphorylation of Drp1 at Serine 616 residue and further recruiting them to mitochondria. NSP4 also promotes mitochondrial translocation of Parkin which reduces mitochondrial fusion by degrading Mfn1. Targeting Drp1 and Cdk1 by respective small molecule inhibitors Mdivi-1 and RO-3306 prevented apoptotic mitochondrial fragmentation and viral progeny release. (f) Subversion of host UPR by RV. Accumulation of misfolded proteins in the ER leads to uncoupling of GRP78 from UPR sensors, resulting in activation of the three branches of UPR-ATF6 pathway, PERK-dependent pathway, and IRE1-based signaling. RV activates two (ATF6 and IRE1) of the three branches of UPR, but limits maturation of the activated UPR pathways. Following RV infection, dissociation of GRP78 from ATF6 (ATF6p90; the transcriptionally inactive fragment) triggers translocation of ATF6 to the Golgi apparatus where it is cleaved and the transcriptionally active fragment ATF6p50 is transported to nucleus to trans-activate UPR elements (CHOP, GADD34, GRP78 and GRP94). Despite the initial activation of ATF6 arm of UPR, RV inhibits further transcription of UPR elements by immobilization of the ATF6p50 fragment into viroplasms. UPR element proteins are also sequestered within viroplasms and further synthesis of them is inhibited by NSP3-induced host translational stasis. Release of PERK from GRP78 leads to homo-dimerization and phosphorylation of PERK; however, RV sequesters p-PERK in the viroplasms inhibiting further activation. Uncoupling of GRP78 from IRE1 leads to homo-dimerization and autophosphorylation of IRE1. Phosphorylated IRE1 (p-IRE1) triggers splicing of xbp1 mRNA (xbp1u) to form a spliced variant (xbp1s). However, further translation of the xbp1s is prevented as a result of general host translational inhibition mediated by RV-NSP3. RV also induces IRE1-independent alternative splicing of xbp1 leading to generation of an exon-skipped splice variant (xbp1es). This event of xbp1 alternative splicing was found to concur with NSP3-mediated PABPC1 nuclear translocation. (g) Evasion of antiviral impacts of SGs and PBs and viroplasmic sequestration of host cellular RBPs. Translational shut off because of phosphorylation of eIF2α is a classical trigger for formation of SGs which accumulate many cellular proteins (see figure). PBs and GW bodies also contain protein conglomerates (see figure). In RV infected cells, eIF2α becomes phosphorylated by PKR, but SG formation is prevented. Punctate PB structures are also absent in infected cells. PB component Pan3 and GW body component AGO2 are degraded proteasomally by RV-NSP1. At later hours, AGO2 relocalizes to viroplasmic niches. Other PB/SG/GW body components are also relocated to different subcellular niches such as nuclear compartment (XRN1, hDcp1, DDX6) or viroplasms (ADAR1, Caprin1, CPEB, eIF2α, PKR, Staufen1, PPM1A, LSM1, PARN, GW182, Caf1) or remained dispersed in cytosol (G3BP1, ZBP1). Moreover, many PB/SG components interact with viroplasmic RV-NSP2 and RV-NSP5. Additionally, many hnRNPs and ARE-BPs are re-located from nucleus to cytosol, get sequestered to viroplasms, and interact with RV-NSP2 and RV-NSP5 within RV infected cells. Many relocated RBPs are also absorbed by the copious viral transcripts
![Figure 3. Evasion of host cellular antiviral responses by RV. (a) Evasion of host cellular IFN response. Within RV infected cells, RV RNA species with exposed 5ʹ-phosphate groups or with incomplete 5ʹ-O-methylated “cap” structures act as PAMPs and are recognized by host cellular PRRs RIG1 and MDA5. Subsequent oligomerization of the mitochondrial adaptor MAVS forms a platform for recruitment of TRAFs and two kinase complexes IKK-α/β/γ and TBK1-IKKε leading to the activation of IRF3/7 and NF-κB-dependent transcriptional programme (IFNs, ISGs, cytokines). NF-κB contains two subunits p50 and p65. IKK-α/β/γ phosphorylates NF-κB inhibitory protein IκB at the α subunit resulting in its proteasomal degradation by SCFβ-TrCP (Skp1-Cullin1-F-box containing protein β-TrCP) E3 ubiquitin ligase complex. Being freed of IκB, p50-p65 heterodimeric NF-κB translocates to nucleus and causes transcriptional activation. RV-NSP1 targets many host proteins of this pathway proteasomally (such as MAVS, β-TrCP, IRF3/7, TRAF2) or non-proteasomally (RIG1) and causes their degradation in a RV strain-dependent manner. RV-VP3 has also been shown to target MAVS for proteasomal degradation. Moreover, at least for β-TrCP degradation, a fostering role of hijacked host cellular Cul3-Rbx1 E3 ubiquitin ligase machinery has been implicated. NSP1-β-TrCP interaction also requires CK-II-directed NSP1 phosphorylation reactions. Some RV strains block NF-κB function through sequestration of NF-κB p65 away from nucleus into viroplasmic puncta. The amplification step of IFN response includes binding of IFNs (type I and type II) to cognate receptors (type I IFN receptor IFNAR and type II IFN receptor IFNGR) to activate JAK-STAT signaling. The phosphorylated forms of STAT1 and STAT2 form a complex called ISGF3 by associating with IRF9. This heterotrimeric complex translocates to the nucleus, binds to IFN-stimulated response elements (ISREs) and trans-activates a series of ISGs of cyto-protective and antiviral nature. Homodimeric phosphorylated STAT1 downstream of IFNGR signaling also trans-activates ISGs by nuclear translocation and binding to Gamma interferon activation site (GAS). RV infection curtails IFN amplification pathway by promoting degradation of IRF9, IFN receptors, and by preventing STAT1 phosphorylation as well as STAT1-STAT2 nuclear translocation. RV-NSP1 is responsible for degradation of IRF9 proteasomally and for inhibition of STAT1 phosphorylation. (b) Evasion of host cellular OAS/RNase L pathway by RV. Within virus infected cells, viral dsRNA population can induce oligomerization-dependent activation of the enzyme OAS which further catalyzes the formation of 2ʹ-5ʹAs. Upon interacting with 2ʹ-5ʹAs, RNase L gets activated through dimerization and triggers cleavage of RNA including the viral RNA species. RV evades the deleterious effects of OAS/RNase L pathway by its structural protein VP3. RV-VP3 has intrinsic 2ʹ, 5ʹ-phosphodiesterase (2ʹ-5ʹPDE) motif through which it disintegrates 2ʹ-5ʹA structures, thereby preventing RNase L activation and viral RNA cleavage. (c) Evasion of innate antiviral impacts of host RNAi machinery by RV. Double stranded RV replication intermediates can potentially be subjected to trimming by host cellular DICER resulting in production of virus‐derived small interfering RNAs (viRNAs). Incorporation of viRNAs into the RISC containing the catalytic effector AGO2 can subsequently target viral RNA population. During early hours of RV-SA11 and RV-A5-13 infection, RV-NSP1 interacts with and ubiquitylates AGO2 leading to proteasomal demise of this catalytic effector. In this respect, RV-NSP1 acts as a putative viral‐suppressor‐of‐RNAi (VSR). In absence of AGO2, siRNA/shRNA-guided RNAi and also potential viRNA-directed RNAi are rendered nonfunctional during early hours of RV infection. Clonal overexpression of AGO2 shows anti-RV effects. (d) Attenuation of host cellular anti-oxidant defense system by RV. Nrf2 is the master transcription factor which deals with cellular redox stress by transcribing anti-oxidant and cyto-protective effectors such as HO-1, NQO1, SOD1. Under unstressed condition, Nrf2 is constantly turned over in a ubiquitin-proteasome-dependent way by cellular Keap1-Rbx1-Cul3 machinery. (Left panel) In RV infected cells, reactive oxygen species (ROS) is induced during early hours leading to Keap1 inhibition and Nrf2 upregulation. Elevated Nrf2, further primed by PKC-mediated phosphorylation, translocates to nucleus and trans-activate stress responsive genes which contain Nrf2-binding motif [anti-oxidant response element (ARE)] in their promoter regions. Quenching ROS by NAC has an antagonizing effect on RV infection. (Right panel) During later hours of RV infection, Nrf2 is expelled out of the nucleus, ubiquitylated by a non-canonical E3 ubiquitin ligase (other than the canonical Keap1-Rbx1-Cul3 machinery) and degraded proteasomally. Levels of HO-1, NQO1, and SOD1 were also reduced. Agonists of Nrf2/ARE pathway, such as Keap1 inhibitors CDDO-Me and RA-839, show potent anti-RV effects. (e) Time-dependent regulation of host cellular apoptotic cell death by RV. (Left panel) During early hours of infection, anti-apoptotic pathways are activated and pro-apoptotic pathways are inhibited for ensuring viral replication. The prime most survival pathway includes activation of PI3K-Akt signaling as a result of interaction of RV-NSP1 with PI3K. Interaction of the chaperone Hsp90 with Akt has an agonistic effect on this pathway. Inhibition of survival pathways through targeting PI3K, phospho-Akt and Hsp90 by LY, 294–002, triciribine and 17-AAG, respectively, sensitized RV replication. Inhibition of pro-apoptotic pathways is multifaceted. One of them is the upregulation of the miRNA population hsa-miR-142-5p by RV-NSP5. Elevated hsa-miR-142-5p sensitizes its targets TGFβR II and SMAD3 leading to attenuation of p38MAPK-ERK1/2-JNK-dependent apoptotic signaling in HT29 cell line. Another strategy is the ubiquitylation and proteasomal degradation of p53 during early infection period. RV-NSP1 plays a pivotal role in this regulation. In absence of p53, transcription of p53-dependent apoptotic genes is prevented. Yet another anti-apoptotic modality in early hours of RV infected cells is the prevention of RV-NSP4 translocation to mitochondria. This is enabled by the ubiquitin-proteasome-dependent demise of the mitochondrial chaperonin Hsp60 which facilitates NSP4 mitochondrial import. A phosphorylation event of Hsp60 carried out by the autophosphorylated and activated form of Src kinase (SrcY416) imparts proteasomal sensitivity to Hsp60. Targeting hsa-miR-142-5p by its anti-miR and Src kinase by a small molecule SKI-I exert anti-RV activity. (Right panel) During late phase of infection, apoptotic pathways are activated and/or de-repressed and outweigh the survival pathways. Intrinsic pathway of apoptosis observed in late hours of RV infected cells is partially dependent on Bax. Subsequent release of cytochrome c into cytosol results in apoptosome formation and activation of executioner caspases. Reduced level of hsa-miR-142-5p results in de-repression of TGFβR II-SMAD3-p38MAPK-ERK1/2-JNK-dependent apoptotic signaling in HT29 cell line. Weakened interaction of p53 with RV-NSP1 stabilizes p53 and causes p53-dependent transcription of pro-apoptotic genes (PUMA, Bax. Bak). In absence of SrcY416, Hsp60 is no longer phosphorylated and therefore escorts NSP4 across mitochondria. NSP4 also positively regulates apoptotic mitochondrial fragmentation by promoting Cdk1-dependent phosphorylation of Drp1 at Serine 616 residue and further recruiting them to mitochondria. NSP4 also promotes mitochondrial translocation of Parkin which reduces mitochondrial fusion by degrading Mfn1. Targeting Drp1 and Cdk1 by respective small molecule inhibitors Mdivi-1 and RO-3306 prevented apoptotic mitochondrial fragmentation and viral progeny release. (f) Subversion of host UPR by RV. Accumulation of misfolded proteins in the ER leads to uncoupling of GRP78 from UPR sensors, resulting in activation of the three branches of UPR-ATF6 pathway, PERK-dependent pathway, and IRE1-based signaling. RV activates two (ATF6 and IRE1) of the three branches of UPR, but limits maturation of the activated UPR pathways. Following RV infection, dissociation of GRP78 from ATF6 (ATF6p90; the transcriptionally inactive fragment) triggers translocation of ATF6 to the Golgi apparatus where it is cleaved and the transcriptionally active fragment ATF6p50 is transported to nucleus to trans-activate UPR elements (CHOP, GADD34, GRP78 and GRP94). Despite the initial activation of ATF6 arm of UPR, RV inhibits further transcription of UPR elements by immobilization of the ATF6p50 fragment into viroplasms. UPR element proteins are also sequestered within viroplasms and further synthesis of them is inhibited by NSP3-induced host translational stasis. Release of PERK from GRP78 leads to homo-dimerization and phosphorylation of PERK; however, RV sequesters p-PERK in the viroplasms inhibiting further activation. Uncoupling of GRP78 from IRE1 leads to homo-dimerization and autophosphorylation of IRE1. Phosphorylated IRE1 (p-IRE1) triggers splicing of xbp1 mRNA (xbp1u) to form a spliced variant (xbp1s). However, further translation of the xbp1s is prevented as a result of general host translational inhibition mediated by RV-NSP3. RV also induces IRE1-independent alternative splicing of xbp1 leading to generation of an exon-skipped splice variant (xbp1es). This event of xbp1 alternative splicing was found to concur with NSP3-mediated PABPC1 nuclear translocation. (g) Evasion of antiviral impacts of SGs and PBs and viroplasmic sequestration of host cellular RBPs. Translational shut off because of phosphorylation of eIF2α is a classical trigger for formation of SGs which accumulate many cellular proteins (see figure). PBs and GW bodies also contain protein conglomerates (see figure). In RV infected cells, eIF2α becomes phosphorylated by PKR, but SG formation is prevented. Punctate PB structures are also absent in infected cells. PB component Pan3 and GW body component AGO2 are degraded proteasomally by RV-NSP1. At later hours, AGO2 relocalizes to viroplasmic niches. Other PB/SG/GW body components are also relocated to different subcellular niches such as nuclear compartment (XRN1, hDcp1, DDX6) or viroplasms (ADAR1, Caprin1, CPEB, eIF2α, PKR, Staufen1, PPM1A, LSM1, PARN, GW182, Caf1) or remained dispersed in cytosol (G3BP1, ZBP1). Moreover, many PB/SG components interact with viroplasmic RV-NSP2 and RV-NSP5. Additionally, many hnRNPs and ARE-BPs are re-located from nucleus to cytosol, get sequestered to viroplasms, and interact with RV-NSP2 and RV-NSP5 within RV infected cells. Many relocated RBPs are also absorbed by the copious viral transcripts](/cms/asset/423dbd89-0b21-4bc3-8cff-3c1f18eed5b8/kvir_a_1903198_f0003a_oc.jpg)
Figure 3. Continued
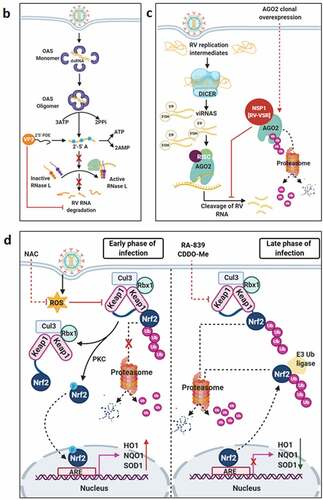
Figure 3. Continued
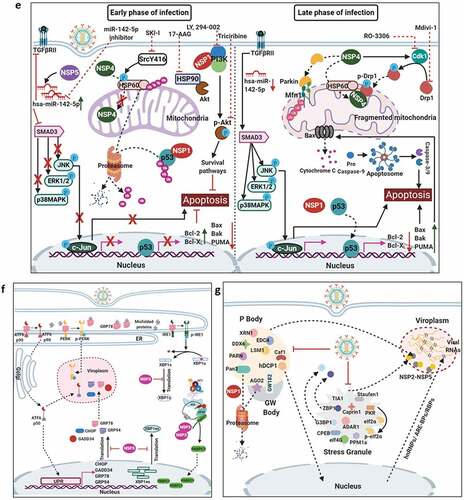
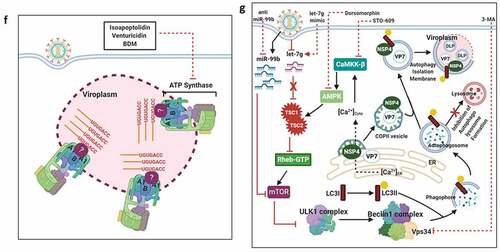
Figure 4. Usurpation of host machineries by RV. (a) Preferential synthesis of viral proteins at the expense with host protein translation. The eukaryotic initiation factor 2 (eIF2) is needed to form the ternary complex (TC) [consisting of eIF2 (α, β, and γ subunits), Met-tRNAi (initiator tRNA carrying methionine), and GTP], which in turn escorts the initiator Met-tRNAi to the P site of the 40S ribosomal subunit, enabling translation initiation. The delivery of the initiating amino acid requires GTP hydrolysis. The guanine exchange factor eIF2B is needed for the conversion of eIF2-GDP to eIF2-GTP for translation to continue. In response to RV infection, the α subunit of eIF2 becomes phosphorylated by the viral dsRNA-activated PKR. The p-eIF2α sequesters eIF2B in an inhibitory complex, leading to prevention of ternary complex formation and global translation inhibition. Efficient translation of eukaryotic mRNA also requires mRNA circularization through interaction between eIF4E (cap-binding protein at the 5ʹ termini) and PABPC1 [Poly(A) tail binding protein at the 3ʹ termini] via an intermediate scaffold eIF4G. During RV infection, NSP3 interacts with eIF4G thereby evicting PABPC1 from PABPC1-eIF4G complex; NSP3 also promotes nuclear re-localization of PABPC1 by interacting with a cellular protein RoXaN. Nuclear aggregation of PABPC1 causes accumulation and hyperadenylation of poly(A)-containing mRNAs because of abrogated nucleus-to-cytoplasmic mRNA export. In times of host translational stasis, rotaviral transcripts are translated on host cellular polysomes to produce viral proteins. (b) Regulation of host calcium metabolism and cell cycle progression by RV. Within virus infected cells, RV-NSP4 acts as a viroporin on ER membrane and releases Ca2+ from ER. Loss of ER Ca2+ activates ER-resident Ca2+-sensor STIM1 which translocate to plasma membrane to further activate calcium channels such as Orai1 and TRPC. Other calcium channels such as sodium/calcium exchanger (NCX) and voltage-activated Ca2+ channels (VACC) also regulate Ca2+ homeostasis; contribution of VACC, however, has been questioned. Several Ca2+ channel blockers, which sensitize NSP4-mediated calcium entry, are shown. Among the signaling cascades activated downstream of increased cytosolic Ca2+ are the Ca2+/CaM/CaMKI/Cyclin-Cdk/Rb/E2F signaling axis to facilitate G1-to-S phase transition of host cells (and the autophagic signaling to aid in outer capsid assembly of progeny virions; omitted here for clarity; shown in detail later). RV-VP6 also interacts directly with CaM in a Ca2+-dependent way. Ca2+ chelation by BAPTA-AM has a negative impact on Ca2+-induced signaling pathways in RV infected cells and reduces RV progeny yield. Moreover, targeting CaM by a small molecule inhibitor W7 also abrogated CaM/CaMKI/Cyclin-Cdk/Rb/E2F signaling axis and antagonized RV progeny production. Besides G1-to-S phase transition, prolonged intra-S phase retention of RV-infected cells is also enabled by a blockade of entry into M phase by cell cycle arrest at S-G2 check point. This is accomplished by the depletion of cyclin B1 and subsequent inhibition of the Cdk1-cyclin B1 complex. RV proteins NSP3, NSP5, and VP2 regulate cell cycle arrest at S-G2 check point. Inhibition of mitotic entry ensures preservation of hyperacetylated and stabilized microtubular structures which along with the kinesin motor protein Eg5 facilitate viroplasmic condensation and peri-nuclear relocalization. Targeting microtubule and Eg5 kinesin by small molecules nocodazole and monastrol, respectively, impairs viroplasm dynamics. Secreted extracellular NSP4 binds to integrins on neighboring uninfected cells and activates PLC-IP3-dependent signaling cascade to elevate intracellular Ca2+ which in turn triggers Ca2+-dependent Cl− secretion through calcium-activated chloride channels (CaCC) such as TMEM16A. Blockers of CaCC, which prevent Ca2+-dependent Cl− secretion, are also shown. Increased cytosolic Ca2+ in EC cells cause granular release of 5-HT which stimulates ENS to release VIP from nerve endings adjacent to crypt cells. VIP triggers increased water and Cl− secretion from crypt cells. 5-HT receptor blockers and VIP receptor antagonists attenuate RV-induced diarrheagenic response. (c) Usurping host cellular lipid droplets for rotaviral viroplasm assembly. LD biogenesis occurs from within ER and precedes sequential enzymatic reactions. Synthesis of fatty acid palmitate from acetyl-CoA and malonyl-CoA requires the FASN complex. The enzyme ACC-1 catalyzes the conversion of acetyl-CoA into malonyl-CoA. Alternatively, the enzyme ACSL converts the fatty acids which are transported across the plasma membrane from the extracellular source into corresponding fatty acyl-CoA esters. Subsequent synthesis of triacylglycerols (TAG) from fatty acid palmitate and sterol esters (SE) from cholesterol requires the ER-localized enzymes DGAT1/DGAT2, and ACAT1/ACAT2, respectively. These neural lipids accumulate within the lipid bilayers of the ER and get incorporated within the nascent LDs which bud from the outer leaflet of the ER into the cytosol. LDs gradually acquire signature protein markers (such as ADRP and Perilipins). In RV infected cells, LDs act as scaffolds for viroplasm formation and maturation. The nucleation step of viroplasms involves two non-structural RV proteins NSP2 and NSP5. Autophosphorylated and cytoplasmically dispersed NSP2 (dNSP2) interacts with hypophosphorylated NSP5 to finally result in viroplasm formation through a series of phosphorylation reactions on NSP5 (thereby forming hyperphosphorylated NSP5) by NSP2 kinase activity and a host cellular kinase CK1α. Within RV infected cells, ADRP, Perilipin A, and CK1α co-localize with viroplasmic NSP2 (vNSP2) and NSP5. Different enzymes of neutral lipid biosynthesis pathway can be targeted with small molecules to result in inhibition of viroplasm formation/maturation and reduced RV progeny production; ACAT1/2 inhibition by CI-976, PHB; ACSL inhibition by Triascin C; ACC-1 inhibition by TOFA; FASN inhibition by C75; DGAT1/2 inhibition by Betulinic acid, A922500. Similarly, silencing CK1α by siRNA leads to impairment of viroplasm dynamicity and RV progeny yield. (d) Interfering with the host cellular nucleotide biosynthesis pathway sensitizes RV infection. Purine biosynthesis de novo (coded in sky blue color) requires a series of enzymatic reactions in which one of the steps is oxidation of inosinate (IMP) to form xanthylate (XMP). This reaction is catalyzed by the enzyme IMP dehydrogenase (IMPDH) which has two isoforms IMPDH1 and IMPDH2. MPA inhibits de novo purine biosynthesis by targeting IMPDH2 and exerts anti-RV effects. Similarly, pyrimidine biosynthesis de novo (coded in light green color) includes two intermediate steps: oxidation of dihydroorotate to orotate by DHODH and formation of uridylate (UMP) from orotidylate (OMP) by ODcase. Targeting DHODH by small molecules such as BQR, LFM and ODcase by 6-AU impairs de novo pyrimidine biosynthesis and exerts anti-RV effects. Anti-RV impacts of gemcitabine depends on inhibition of salvage pathway of pyrimidine biosynthesis (coded in pink color). (e) Usurping a non-canonical DDR signaling by RV to regulate viroplasm dynamicity. (Lower panel) In RV infected cells, MRN-ATM-Chk2 branch of DDR is activated in absence of nuclear DNA damage by an unknown RV-induced mechanism. Formation of γ-H2AX-positive nuclear foci is also prevented. MRN components, ATM, and Chk2 are translocated from nucleus to cytopolasm and accumulate within viroplasmic niches to regulate viroplasm dynamicity. Targeting Mre11, ATM, and Chk2 by small molecule inhibitors Mirin, KU55933, and BML-277, respectively, antagonize RV replication. KU55933 and BML-277 prevent viroplasm condensation and maturation. (Upper panel) However, enforcing DNA damage externally by etoposide or by disintegrating nuclear Cohesin complex (because of loss of STAG2) causes nuclear induction of γ-H2AX and cytosolic accumulation of DNA leading to the activation of cGAS-cGAMP-STING-TBK1-IRF3 signaling. IRF3-dependent IFN synthesis and subsequent amplification of JAK-STAT-mediated antiviral IFN response lead to a RV-refractory host cellular state; cGAMP (2′3′-Cyclic GMP-AMP) (f) Co-opting host cellular ATP synthase for rotaviral life cycle. Three subunits of mitochondrial ATP synthase holoenzyme (ATP5B, ATP5A1, ATP5O) are re-located to viroplasms and interact with 3ʹ UTR consensus of RV RNAs (5ʹ-UGUGACC-3ʹ). Potential involvement of an intermediate viral protein (such as VP1) has been speculated to facilitate the ATP synthase-3ʹ UTR association. Chemical inhibitors targeting ATP synthase such as Isoapoptolidin, Venturicidin, and BDM antagonize RV progeny yield. (g) Exploiting host cellular autophagic machinery for rotaviral morphogenesis. Induction of autophagic signaling occurs in RV infected cells by two mutually exclusive triggers. One of the triggers includes two miRNAs, miR-99b and let-7 g, as the pivotal regulatory components. RV infection increases miR-99b population and decreases let-7 g. Increased miR-99b suppresses mTOR which is a direct target of it. Moreover, reduced let-7 g increases its target TSC1 leading to stabilization of TSC1-TSC2 complex. Because of the GTPase activity of TSC2 for Rheb-GTP, TSC1-TSC2 stabilization results in attenuated levels of Rheb-GTP pool. Rheb-GTP being a positive regulator of mTOR, RV-mediated Rheb-GTP depletion antagonizes mTOR. The other stimulus initiates with the cytosolic release of Ca2+ through the NSP4 viroporin activity on ER membrane, followed by the sequential activation of CaMKK-β and AMPK. AMPK directs TSC1-TSC2 stabilization which finally culminates in mTOR inhibition. Overall mTOR restriction causes de-repression of ULK1 complex which subsequently forms phagophore through Beclin1 complex activation and LC3 II lipidation. However, autophagosomes are prevented from lysosomal targeting in RV infected cells; instead, they are utilized for carrying the RV proteins NSP4 and VP7 coming out with the ER-derived COP-II vesicles to maturing progeny virions within viroplasms, thereby aiding in outer capsid assembly. Host regulators that can be targeted for inhibiting autophagy and restricting RV infection are shown; AMPK, CaMKK-β, Vps34 and miR-99b inhibition by Dorsomorphin, STO-609, 3-MA, and anti-miR-99b, respectively; let-7 g elevation by let-7 g mimic
![Figure 4. Usurpation of host machineries by RV. (a) Preferential synthesis of viral proteins at the expense with host protein translation. The eukaryotic initiation factor 2 (eIF2) is needed to form the ternary complex (TC) [consisting of eIF2 (α, β, and γ subunits), Met-tRNAi (initiator tRNA carrying methionine), and GTP], which in turn escorts the initiator Met-tRNAi to the P site of the 40S ribosomal subunit, enabling translation initiation. The delivery of the initiating amino acid requires GTP hydrolysis. The guanine exchange factor eIF2B is needed for the conversion of eIF2-GDP to eIF2-GTP for translation to continue. In response to RV infection, the α subunit of eIF2 becomes phosphorylated by the viral dsRNA-activated PKR. The p-eIF2α sequesters eIF2B in an inhibitory complex, leading to prevention of ternary complex formation and global translation inhibition. Efficient translation of eukaryotic mRNA also requires mRNA circularization through interaction between eIF4E (cap-binding protein at the 5ʹ termini) and PABPC1 [Poly(A) tail binding protein at the 3ʹ termini] via an intermediate scaffold eIF4G. During RV infection, NSP3 interacts with eIF4G thereby evicting PABPC1 from PABPC1-eIF4G complex; NSP3 also promotes nuclear re-localization of PABPC1 by interacting with a cellular protein RoXaN. Nuclear aggregation of PABPC1 causes accumulation and hyperadenylation of poly(A)-containing mRNAs because of abrogated nucleus-to-cytoplasmic mRNA export. In times of host translational stasis, rotaviral transcripts are translated on host cellular polysomes to produce viral proteins. (b) Regulation of host calcium metabolism and cell cycle progression by RV. Within virus infected cells, RV-NSP4 acts as a viroporin on ER membrane and releases Ca2+ from ER. Loss of ER Ca2+ activates ER-resident Ca2+-sensor STIM1 which translocate to plasma membrane to further activate calcium channels such as Orai1 and TRPC. Other calcium channels such as sodium/calcium exchanger (NCX) and voltage-activated Ca2+ channels (VACC) also regulate Ca2+ homeostasis; contribution of VACC, however, has been questioned. Several Ca2+ channel blockers, which sensitize NSP4-mediated calcium entry, are shown. Among the signaling cascades activated downstream of increased cytosolic Ca2+ are the Ca2+/CaM/CaMKI/Cyclin-Cdk/Rb/E2F signaling axis to facilitate G1-to-S phase transition of host cells (and the autophagic signaling to aid in outer capsid assembly of progeny virions; omitted here for clarity; shown in detail later). RV-VP6 also interacts directly with CaM in a Ca2+-dependent way. Ca2+ chelation by BAPTA-AM has a negative impact on Ca2+-induced signaling pathways in RV infected cells and reduces RV progeny yield. Moreover, targeting CaM by a small molecule inhibitor W7 also abrogated CaM/CaMKI/Cyclin-Cdk/Rb/E2F signaling axis and antagonized RV progeny production. Besides G1-to-S phase transition, prolonged intra-S phase retention of RV-infected cells is also enabled by a blockade of entry into M phase by cell cycle arrest at S-G2 check point. This is accomplished by the depletion of cyclin B1 and subsequent inhibition of the Cdk1-cyclin B1 complex. RV proteins NSP3, NSP5, and VP2 regulate cell cycle arrest at S-G2 check point. Inhibition of mitotic entry ensures preservation of hyperacetylated and stabilized microtubular structures which along with the kinesin motor protein Eg5 facilitate viroplasmic condensation and peri-nuclear relocalization. Targeting microtubule and Eg5 kinesin by small molecules nocodazole and monastrol, respectively, impairs viroplasm dynamics. Secreted extracellular NSP4 binds to integrins on neighboring uninfected cells and activates PLC-IP3-dependent signaling cascade to elevate intracellular Ca2+ which in turn triggers Ca2+-dependent Cl− secretion through calcium-activated chloride channels (CaCC) such as TMEM16A. Blockers of CaCC, which prevent Ca2+-dependent Cl− secretion, are also shown. Increased cytosolic Ca2+ in EC cells cause granular release of 5-HT which stimulates ENS to release VIP from nerve endings adjacent to crypt cells. VIP triggers increased water and Cl− secretion from crypt cells. 5-HT receptor blockers and VIP receptor antagonists attenuate RV-induced diarrheagenic response. (c) Usurping host cellular lipid droplets for rotaviral viroplasm assembly. LD biogenesis occurs from within ER and precedes sequential enzymatic reactions. Synthesis of fatty acid palmitate from acetyl-CoA and malonyl-CoA requires the FASN complex. The enzyme ACC-1 catalyzes the conversion of acetyl-CoA into malonyl-CoA. Alternatively, the enzyme ACSL converts the fatty acids which are transported across the plasma membrane from the extracellular source into corresponding fatty acyl-CoA esters. Subsequent synthesis of triacylglycerols (TAG) from fatty acid palmitate and sterol esters (SE) from cholesterol requires the ER-localized enzymes DGAT1/DGAT2, and ACAT1/ACAT2, respectively. These neural lipids accumulate within the lipid bilayers of the ER and get incorporated within the nascent LDs which bud from the outer leaflet of the ER into the cytosol. LDs gradually acquire signature protein markers (such as ADRP and Perilipins). In RV infected cells, LDs act as scaffolds for viroplasm formation and maturation. The nucleation step of viroplasms involves two non-structural RV proteins NSP2 and NSP5. Autophosphorylated and cytoplasmically dispersed NSP2 (dNSP2) interacts with hypophosphorylated NSP5 to finally result in viroplasm formation through a series of phosphorylation reactions on NSP5 (thereby forming hyperphosphorylated NSP5) by NSP2 kinase activity and a host cellular kinase CK1α. Within RV infected cells, ADRP, Perilipin A, and CK1α co-localize with viroplasmic NSP2 (vNSP2) and NSP5. Different enzymes of neutral lipid biosynthesis pathway can be targeted with small molecules to result in inhibition of viroplasm formation/maturation and reduced RV progeny production; ACAT1/2 inhibition by CI-976, PHB; ACSL inhibition by Triascin C; ACC-1 inhibition by TOFA; FASN inhibition by C75; DGAT1/2 inhibition by Betulinic acid, A922500. Similarly, silencing CK1α by siRNA leads to impairment of viroplasm dynamicity and RV progeny yield. (d) Interfering with the host cellular nucleotide biosynthesis pathway sensitizes RV infection. Purine biosynthesis de novo (coded in sky blue color) requires a series of enzymatic reactions in which one of the steps is oxidation of inosinate (IMP) to form xanthylate (XMP). This reaction is catalyzed by the enzyme IMP dehydrogenase (IMPDH) which has two isoforms IMPDH1 and IMPDH2. MPA inhibits de novo purine biosynthesis by targeting IMPDH2 and exerts anti-RV effects. Similarly, pyrimidine biosynthesis de novo (coded in light green color) includes two intermediate steps: oxidation of dihydroorotate to orotate by DHODH and formation of uridylate (UMP) from orotidylate (OMP) by ODcase. Targeting DHODH by small molecules such as BQR, LFM and ODcase by 6-AU impairs de novo pyrimidine biosynthesis and exerts anti-RV effects. Anti-RV impacts of gemcitabine depends on inhibition of salvage pathway of pyrimidine biosynthesis (coded in pink color). (e) Usurping a non-canonical DDR signaling by RV to regulate viroplasm dynamicity. (Lower panel) In RV infected cells, MRN-ATM-Chk2 branch of DDR is activated in absence of nuclear DNA damage by an unknown RV-induced mechanism. Formation of γ-H2AX-positive nuclear foci is also prevented. MRN components, ATM, and Chk2 are translocated from nucleus to cytopolasm and accumulate within viroplasmic niches to regulate viroplasm dynamicity. Targeting Mre11, ATM, and Chk2 by small molecule inhibitors Mirin, KU55933, and BML-277, respectively, antagonize RV replication. KU55933 and BML-277 prevent viroplasm condensation and maturation. (Upper panel) However, enforcing DNA damage externally by etoposide or by disintegrating nuclear Cohesin complex (because of loss of STAG2) causes nuclear induction of γ-H2AX and cytosolic accumulation of DNA leading to the activation of cGAS-cGAMP-STING-TBK1-IRF3 signaling. IRF3-dependent IFN synthesis and subsequent amplification of JAK-STAT-mediated antiviral IFN response lead to a RV-refractory host cellular state; cGAMP (2′3′-Cyclic GMP-AMP) (f) Co-opting host cellular ATP synthase for rotaviral life cycle. Three subunits of mitochondrial ATP synthase holoenzyme (ATP5B, ATP5A1, ATP5O) are re-located to viroplasms and interact with 3ʹ UTR consensus of RV RNAs (5ʹ-UGUGACC-3ʹ). Potential involvement of an intermediate viral protein (such as VP1) has been speculated to facilitate the ATP synthase-3ʹ UTR association. Chemical inhibitors targeting ATP synthase such as Isoapoptolidin, Venturicidin, and BDM antagonize RV progeny yield. (g) Exploiting host cellular autophagic machinery for rotaviral morphogenesis. Induction of autophagic signaling occurs in RV infected cells by two mutually exclusive triggers. One of the triggers includes two miRNAs, miR-99b and let-7 g, as the pivotal regulatory components. RV infection increases miR-99b population and decreases let-7 g. Increased miR-99b suppresses mTOR which is a direct target of it. Moreover, reduced let-7 g increases its target TSC1 leading to stabilization of TSC1-TSC2 complex. Because of the GTPase activity of TSC2 for Rheb-GTP, TSC1-TSC2 stabilization results in attenuated levels of Rheb-GTP pool. Rheb-GTP being a positive regulator of mTOR, RV-mediated Rheb-GTP depletion antagonizes mTOR. The other stimulus initiates with the cytosolic release of Ca2+ through the NSP4 viroporin activity on ER membrane, followed by the sequential activation of CaMKK-β and AMPK. AMPK directs TSC1-TSC2 stabilization which finally culminates in mTOR inhibition. Overall mTOR restriction causes de-repression of ULK1 complex which subsequently forms phagophore through Beclin1 complex activation and LC3 II lipidation. However, autophagosomes are prevented from lysosomal targeting in RV infected cells; instead, they are utilized for carrying the RV proteins NSP4 and VP7 coming out with the ER-derived COP-II vesicles to maturing progeny virions within viroplasms, thereby aiding in outer capsid assembly. Host regulators that can be targeted for inhibiting autophagy and restricting RV infection are shown; AMPK, CaMKK-β, Vps34 and miR-99b inhibition by Dorsomorphin, STO-609, 3-MA, and anti-miR-99b, respectively; let-7 g elevation by let-7 g mimic](/cms/asset/0f0a6a02-e3b8-49a3-b7d5-39bda296fd68/kvir_a_1903198_f0004a_oc.jpg)
Figure 4. Continued
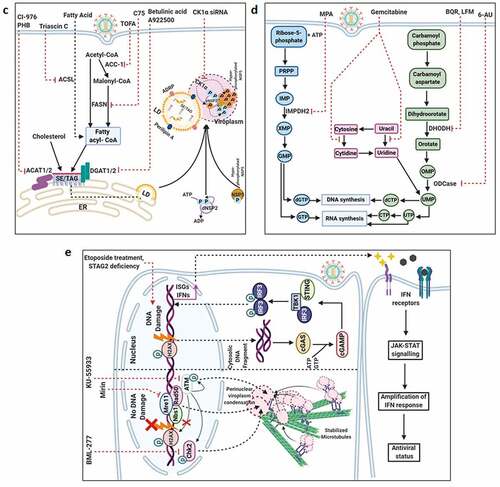
Figure 4. Continued