
Figures & data
Figure 1. The structure of the HBV genome. The locations of the regulatory elements including EN1 and EN2 enhancers and the four promoters are shown. The HBV RNA transcripts, their approximate transcription initiation sites, and the unique poly(A) site are also indicated. The locations of the four ORFs are shown. The unique EcoRI cutting site is defined as nucleotide (nt.) 1. The nt. number is based on the HBV adw2 isolate (genotype A). Cp, core promoter; PreS1p, preS1 promoter; Sp, S promoter; Xp, X promoter.
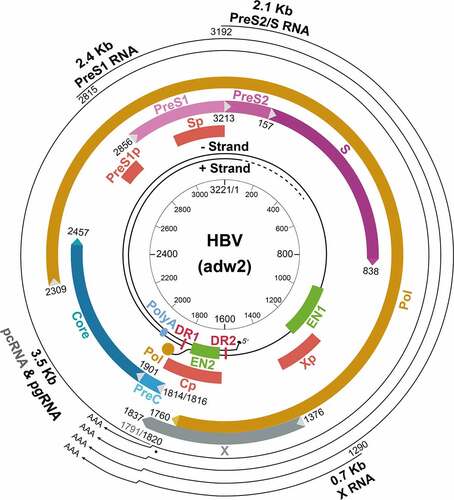
Table 1. Hepatocyte-enriched transcription factors that regulate HBV promoters and enhancers
Figure 2a. Schematic illustration of HBx functional domains. The functional domains of HBx that had been reported are shown.
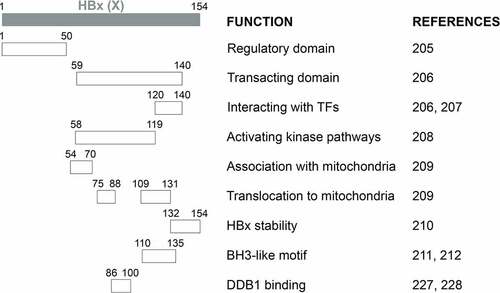
Figure 2b. Continued.
Figure 3. Biological activities of HBx. HBx regulates multiple cellular pathways. It can interact with many transcription factors to activate the expression of host genes, such as MHC class I, iNOS, and IL-8. Its regulation of the CREB-miR-3188 pathway may also play a role in the induction of hepatocarcinogenesis. HBx also epigenetically upregulates HBV gene expression by interacting with a methyltransferase, PRMT1. In addition, HBx suppresses the proteasomal function but is also degraded by the proteasome. It also interacts with the E3 ubiquitin ligase complex to promote the degradation of Smc5/6 and the activation of HBV gene expression. In addition, by interacting with the transcription factor TFIIH, HBx interferes with the host DNA repair machinery, resulting in the accumulation of host DNA damages. Finally, HBx can also activate cellular kinase signaling transduction pathways, including the interaction with mitochondria and the modulation of cytosolic Ca2+ levels to activate the Pyk2-Src pathway, and the activation of the Ras-Raf-MAPK pathway. The involvement of HBx in other pathways such as apoptosis, cell cycles, epigenetic signaling regulations, and autophagy is not illustrated in the figure.
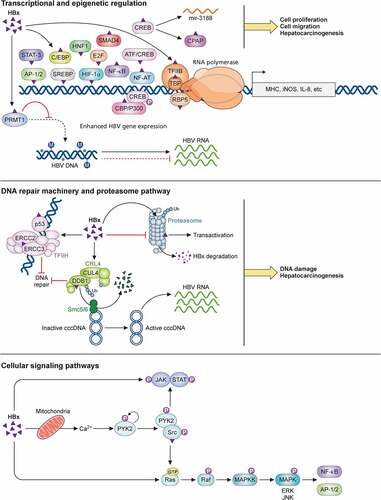
Figure 4. Illustration of the HBV life cycle. HBV initiates its infection of hepatocytes by binding to its receptor NTCP on the cell surface, with the assistance of EGFR. This results in the internalization of the viral particle via a pathway that is still unresolved. The nucleocapsid is subsequently released from the internalized membrane vesicles and transported to the nucleus in a process that involves microtubules. The nuclear transporter factor importin α/β then guides the capsid particle into the nuclear basket, where the HBV genome is released to the nucleoplasm. The partially double-stranded HBV genomic DNA (rcDNA) is converted to cccDNA, possibly by the host DNA repair mechanism, and then forms a mini-chromosome with the addition of nucleosomes, the HBV core protein and HBx. This mini-chromosome directs the synthesis of 3.5-kb pcRNA and pgRNA, 2.4-kb preS1 mRNA, 2.1-kb preS2/S mRNA, and 0.7-kb X mRNA, which serve as the templates for the synthesis of HBeAg, core proteins, polymerase, L, M, and S HBsAg proteins, and HBx, respectively. The pgRNA is packaged together with the DNA polymerase by the core protein to form the nucleocapsid. The reverse transcription ensues to convert the pgRNA into the rcDNA genome. The nucleocapsid may deliver the rcDNA back into the nucleus for the amplification of cccDNA. Alternatively, it may interact with HBsAg for envelopment and the formation of the mature HBV particle. Both multivesicular bodies (MVBs) and endosomal sorting complexes required for transport (ESCRT) are involved in the envelopment and the egress of HBV virions. In addition to complete virions, subviral particles (SVPs) consisting solely of HBsAg, empty virions, RNA-containing virions, and empty capsid particles are also released from HBV-infected cells.
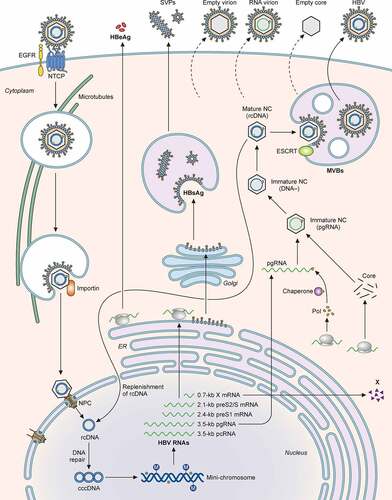
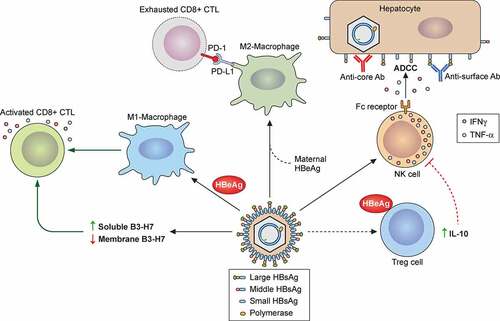
Figure 5. Roles of immune cells in HBV-induced liver pathogenesis. HBV particles and HBeAg can activate hepatic macrophages (Kupffer cells) to undergo the pro-inflammatory M1 polarization, which subsequently express TNF-α and IL-1β and stimulate HBV-specific CTLs for HBV clearance. Alternatively, HBV in the presence of HBeAg can also stimulate hepatic macrophages that have been conditioned by maternal HBeAg to undergo the anti-inflammatory M2 polarization with an increased expression of PD-L1. The binding of PD-L1 to PD-1 on HBV-specific CTLs can lead to T cell exhaustion and HBV persistence. In chronic HBV patients, an elevated level of B7-H3 in the plasma can impair the activity of HBV-specific CTLs. HBV replication can activate NKs to produce IFN-γ and TNF-α to suppress HBV replication. Moreover, the Fc receptor of NKs can recognize anti-surface or anti-core antibodies bound to the surface of HBV-infected hepatocytes to trigger the ADCC during acute and chronic HBV infection. HBeAg can also stimulate Tregs to express IL-10 to cause the dysfunction of NK cells and the establishment of immune tolerance during chronic HBV infection.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this review.