
Figures & data
Figure 1. (a) Circular phylogenetic analysis of the complete genomes of Shigella: Phylogenetic tree showing the relationships of genomes of a total 134 Shigella strains including an Antarctica isolate Shigella sp. PAMC28760 (represented in red text), and their phylogenetic position. This analysis was prepared using MEGA X based on 16S rRNA sequences with neighbour-joining method with 1,000-replicate bootstrap. (b) Heatmap generated with OrthoANI values calculated using the OAT software to determine the close relationship of strain S. sp. PAMC28760 with S. flexneri ATCC29903(T), S. sonnei CECT4887(T), E. coli ATCC11775(T), S. boydii GTC779(T), E. fergusonii ATCC35469(T), S. dysenteriae ATCC13313(T), and E. albertii TW07627(T).
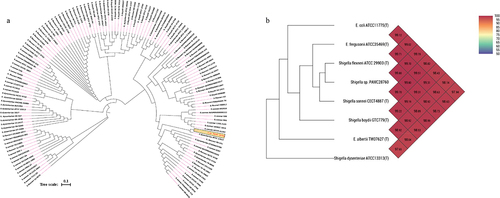
Figure 2. Circular genome comparison using CGView ServerBETA (http://cgview.Ca/) tool for the representation of genome and features of the S. sp. PAMC28760. The contents of the featured rings (starting with the outermost ring to the centre) are as follows. Ring 1, combined ORFs in forward and reverse strands; Ring 2, trehalose degradative genes, combined forward and reverse strand, and CDS (including tRNA and rRNA) in forward and reverse strands; Ring 3, GC skew plot, values above average are depicted in green, and below average in purple; Ring 4, GC content plot; and Ring 5, Sequence ruler.
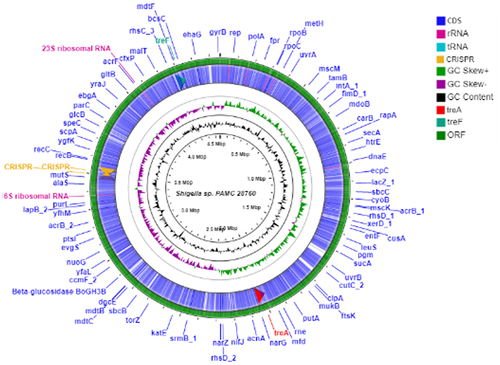
Figure 3. Cytoplasmic trehalase (TreF) amino acid sequence alignment with a characterized trehalase (TreF). TreF (GH37) from E. coli K-12 substr. MG1655, trehalase from S. flexneri C32, trehalase from Shigella sp. PAMC28760, and trehalase from S. boydii ATCC49812. The signature motif 1 and signature motif 2 represent two highly conserved sequence segments that belong to the GH37 family. The “#” symbol denotes the catalytic sites of Asp312 and Glu496. the three black boxes represent conserved regions (CR3–CR5).

Figure 4. Venn diagram categorizes trehalase genes involved in the complete genomes of four Shigella species along with uncategorized Shigella sp. PAMC28760. Green circle represents the cytoplasmic trehalase (treF), whereas red circle represents the periplasmic trehalase (treA). The number outside the circles represents the absence of both trehalase genes.
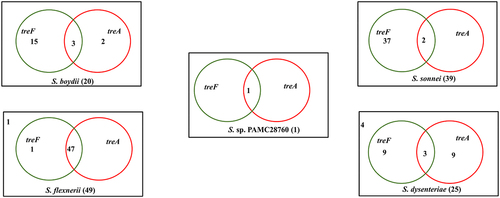
Figure 5. Circular phylogenetic tree based on trehalase genes (treF/treA) sequence in the complete genomes of Shigella strains with reference to the characterized trehalase of E. coli strain K-12 substrain MG165 using a neighbour-joining tree method with 1,000-replicate bootstrap. The pink highlighted boxes represent the characterized trehalase genes (treF and treA), whereas the red text indicates the strain (Shigella sp. PAMC28760) under study.
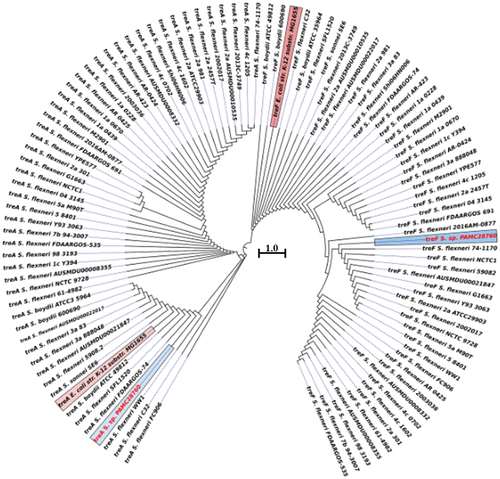
Figure 6. Trehalose degradative pathways. Six different trehalose degradative pathways are found in organisms (bacteria, fungi, yeast, Arthropoda, and plants). Among them, only two degradation pathways (Trehalose degradation pathway II (cytosolic) and VI (periplasmic)) are found in Shigella species.
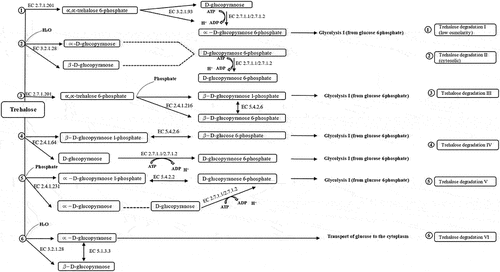
Figure 7. Schematic diagram of the trehalose metabolism pathway in Gram-negative bacteria is formulated from Kosciow et al., 2014 and Purvis et al., 2005. The green boxes represent the trehalose synthesis genes (otsA, trehalose-6-phosphate phosphatase; otsB, trehalose-6-phosphate synthase; and treC, trehalose-6-phosphate hydrolase), whereas grey boxes represent the trehalose degrading genes (treA, periplasmic trehalase; and treF, cytoplasmic trehalase). At cytoplasm, trehalose is degraded by cytoplasmic trehalase gene (treF). The plasma membrane, stretch-activated proteins (SAP) facilitate the exit of trehalose under hypotonic conditions to the periplasm where it further degraded by periplasmic trehalase gene (treA).
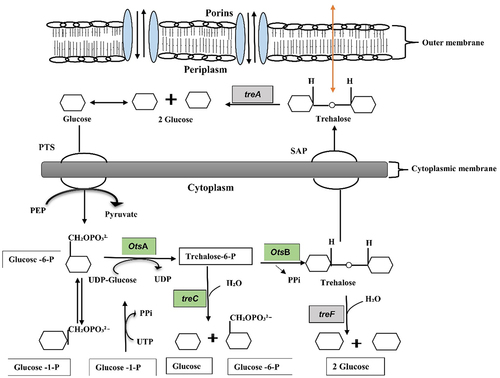
Table 1. MP3 prediction of the total proteins, pathogenic protein, and non-pathogenic proteins in all the complete genomes of Shigella strains including Shigella sp. PAMC28760, which is indicated as a asterisk symbol. Hybrid: predictions from both HMM and SVM models.
Data availability statement
Data used in this study are available from the corresponding author upon reasonable request.