
Figures & data
Figure 1. DEX inhibited OGD/R – mediated myocardial injury.
OGD/R-induced cardiomyocytes (AC16 and HCM) were manipulated with varying concentrations (0.5, 1, and 5 μM) of DEX for 48 hours. A: The viability of each group of cardiomyocytes (AC16 and HCM) was assayed with CCK-8. B: Detection of the proliferation in AC16 and HCM was made using BrdU. C: The profiles of apoptosis-related proteins (Bax, Bcl2, Bad and Caspase3, Caspase8, Caspase9) in AC16 and HCM were determined by WB. D. RT-PCR was conducted to verify the expression of inflammatory factors (TNF-α and IL-1β) in AC16 and HCM. Data were expressed as mean ±SD, n = 3; NS P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (vs. CON). &P < 0.05, &&P < 0.01, &&&P < 0.001 (vs. OGD/R).
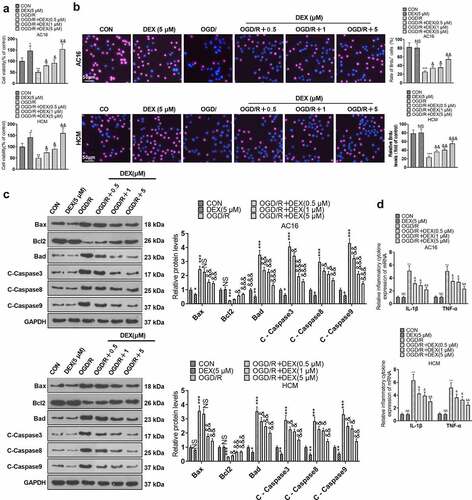
Figure 2. DEX facilitated Sirt1 and inactivated NF-κB.
OGD/R-induced cardiomyocytes (AC16 and HCM) were processed with different concentrations (0.5, 1, and 5 μM) of DEX for 48 hours. A-B: Expression of Sirt1 and NF-κB in AC16 and HCM was assessed by WB. C-D. WB was carried out for detecting the protein expression of Sirt1, NF-κB p65, and AC NF-κB p65 in AC16 and HCM cells transfected with Sirt1 overexpression plasmids or si-Sirt1. Data were expressed as mean ±SD, n = 3; **P < 0.01, ***P < 0.001(vs.CON). &P < 0.05, &&P < 0.01, &&&P < 0.001 (vs. OGD/R).
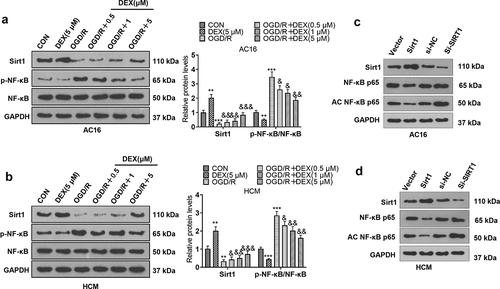
Figure 3. Inhibiting Sirt1 attenuated DEX-mediated myocardial protection.
OGD/R-induced cardiomyocytes (AC16 and HCM) were treated with 5 μM DEX for 48 hours, followed by the addition of a Sirt1 inhibitor (EX527). A: Expression of Sirt1 and NF-κB in AC16 and HCM was determined by WB. B: Detection of cardiomyocyte viability was performed in AC16 and HCM using CCK-8 assay. C: Cardiomyocyte proliferation was checked by the BrdU assay. D: Expression of apoptotic proteins (Bax, Bcl2, Bad and Caspase3, Caspase8, and Caspase9) in AC16 and HCM was monitored by WB. E. RT-PCR was conducted to measure the expression of inflammatory factors (TNF-α and IL-1β) in AC16 and HCM. Data were expressed as mean ±SD, n = 3; **P < 0.01, ***P < 0.001 (vs. OGD/R). &P < 0.05, &&P < 0.01, &&&P < 0.001 (vs. OGD/R+ DEX).
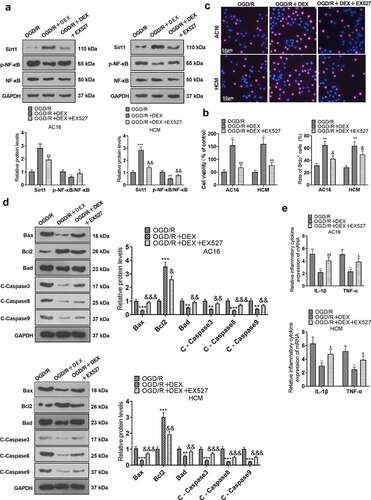
Figure 4. DEX boosted TET1 and abated OGD/R-mediated DNA methylation in cardiomyocytes.
OGD/R-induced cardiomyocytes (AC16 and HCM) were processed with varying concentrations (0.5, 1, and 5 μM) of DEX for 48 hours. A: Expression of TET1 and DNA methylation-related proteins (DNMT1, DNMT3A, and DNMT3B) in AC16 and HCM was tested by WB. B: DNA methylation levels in AC16 and HCM were measured by adopting the 5-mC DNA ELISA Kit. Data were expressed as mean ±SD, n = 3; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (vs. CON). &P < 0.05, &&P < 0.01, &&&P < 0.001 (vs. OGD/R), &&P < 0.01.
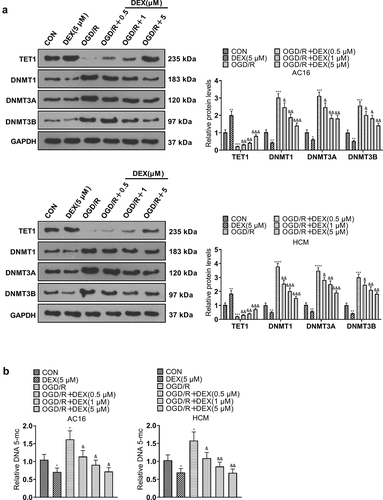
Figure 5. TET1 knockdown impeded the DEX-mediated myocardial protective effect.
The si-NC and si-TET1 were transfected into cardiomyocytes (HCM and AC16), and the transfected OGD/R-induced cardiomyocytes were then treated with 5 μM DEX for 48 hours. A: TET1 expression in AC16 and HCM was checked by WB. B: The viability of AC16 and HCM was tested using CCK-8. C: The BrdU assay was conducted to examine the proliferation of AC16 and HCM. D: The profiles of Bax, Bcl2, Bad, Caspase3, Caspase8, and Caspase9 in AC16 and HCM were determined by WB. E. The profiles of inflammatory factors (TNF-α and IL-1β) in AC16 and HCM were evaluated by RT-PCR. Data were expressed as mean ±SD, n = 3; **P < 0.01, ***P < 0.001 (vs. OGD/R). &P < 0.05, &&P < 0.01, &&&P < 0.001 (vs. OGD/R+ DEX+si-NC).
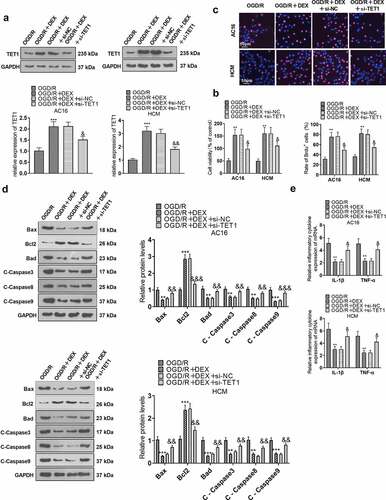
Figure 6. TET1 mediated demethylation of the Sirt1 promoter and lifted Sirt1 expression.
TET1 overexpression plasmids were transfected into cardiomyocytes (HCM and AC16), and then OGD/R-induced cardiomyocytes were processed with 5 μM DEX for 48 hours, followed by the addition of a Sirt1 inhibitor (EX527). A: The levels of TET1, Sirt1, and NF-κB in AC16 and HCM cells were determined by WB. B: AC16 and HCM cells were treated with OGD/R. MSPCR was adopted for checking the Sirt1 promoter methylation in AC16 and HCM. Data were expressed as mean ±SD, n = 3; **P < 0.01, ***P < 0.001 (vs. OGD/R).NS P > 0.05, &&P < 0.01 (vs. OGD/R+ DEX). #P < 0.05, ##P < 0.01, ###P < 0.001 (vs. OGD/R+ DEX+EX527).
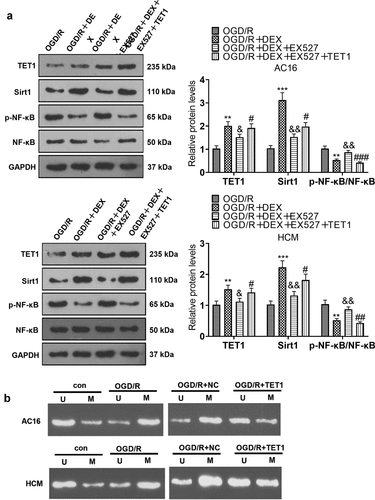
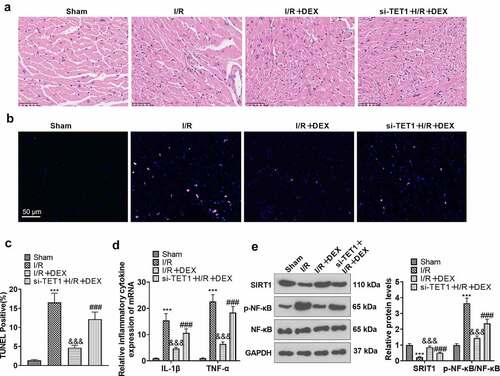
Figure 7. Knockdown of TET1 weakened DEX-mediated myocardial protection.
The rat I/R model was established, and the rats were randomized into Sham, DEX, I/R, I/R + DEX, and si-TET1 + I/R + DEX groups. A. H&E staining was applied to inspect the pathological changes in myocardial tissues of rats. B. TUNEL was employed to gauge apoptosis in rat myocardial tissues. C. The expression of inflammatory factors IL-1β and TNF-α in rat myocardial tissues was tested by RT-PCR. D. The expression of Sirt1 and NF-κB in rat myocardial tissues was monitored by WB. ***P < 0.001 (vs. Sham). &&&P < 0.01 (vs. I/R). ###P < 0.001 (vs. I/R+ DEX). Data were expressed as mean ±SD, n = 5.
Data availability statement
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.