Figures & data
Table 1 The Underlying Mechanisms of OS Induced Depression, as Revealed by Clinical Studies.
Table 2 The Underlying Mechanisms of OS Induced Depression, as Revealed by Rodent Studies.
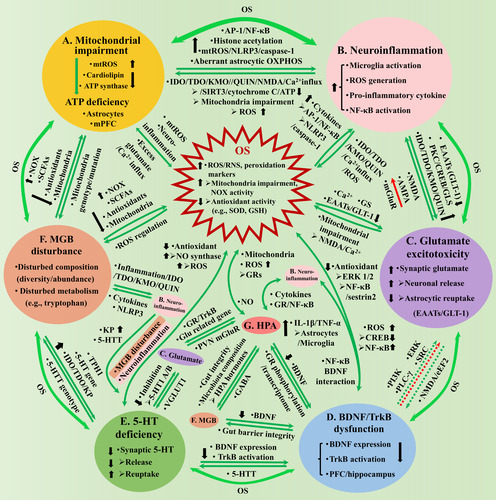
Figure 1 Os causes depression through inducing mitochondrial impairment (A), neuroinflammation (B), glutamate excitotoxicity (C), BDNF/TrkB dysfunction (D) and 5-HT deficiency (E) in the brain, and the MGB disturbance (F) and the HPA axis dysregulation (G). A. Mitochondrial impairment induces central ATP deficiency, through elevating mtROS and reducing cardiolipin and ATP synthase, with astrocytes derived and mPFC localized ATP deficiency closely associated with depression. OS and mitochondria have mutual influence through mtROS, neuroinflammation and excess glutamate induced Ca2+ influx (A-OS). B. Neuroinflammation is characterized by microglia activation, pro-inflammatory cytokine expressions, etc. OS/Mitochondria-neuroinflammation interaction (AB, B-OS): Excess ROS increases pro-inflammatory cytokines, through elevating histone acetylation and activating NLRP3/caspase-1, AP-1 and NF-κB, and aberrant astrocytic OXPHOS activity; neuroinflammation promotes IDO/TDO/KMO to generate QUIN that activates NMDA, increases glutamate, triggers Ca2+ influx, which causes cytochrome C release and reduces SIRT3 activity and ATP generation, or depolarizes mitochondria membrane and generates excess ROS. C. Glutamate excitotoxicity is characterized by central synaptic accumulation. Glutamate-OS interaction (C-OS): Excess ROS results in excitotoxicity through affecting Ca2+, GS and GLT-1 expression; excess glutamate impairs mitochondria through NMDA activation and Ca2+ influx. Neuroinflammation-glutamate interaction (BC): Neuroinflammation promotes excitotoxicity through increasing microglial release or blocking astrocytic reuptake, by downregulating GLT-1 or elevating GLS and QUIN; excess glutamate might increase pro-inflammatory cytokines through NMDA/AMPA receptors, but reduce the cytokines through mGluRs. D. BDNF/TrkB dysfunction is characterized by inhibited BDNF expression and TrkB activation (PFC/hippocampus). BDNF-OS interaction (D-OS): Excess ROS inhibits BDNF expression, through reducing CREB and elevating NF-κB activity; BDNF owns antioxidant property through TrkB, ERK1/2 and NF-κB/sestrin2, thus, the dysfunction promotes OS. Neuroinflammation-BDNF/TrkB interaction (BD): NF-κB mediates LPS-reduced BDNF expression through interacting with BDNF/TrkB; BDNF promotes TNF-α and IL-1β release by activating astrocytes and microglia. Glutamate-BDNF/TrkB interaction (CD): Excess glutamate might inhibit BDNF expression by activating extra-synaptic NMDA and eEF2; BDNF promotes the release or protects neurons against the excitotoxicity of glutamate, through PI3K, PLC-γ, ERK, etc., whose dysfunction weakens the influence correspondingly. E. Central 5-HT interacts with multiple aspects, whose deficiency attenuates the influence correspondingly: 5-HT presents antioxidant properties, while OS inhibits 5-HT through neuroinflammation and MGB disturbance (E-OS); Neuroinflammation inhibits 5-HT by promoting KP and modulating 5-HTT (BE); 5-HT inhibits glutamate transmission through 5-HT 1A/B receptor, which in turn regulates 5-HT function through VGLUT1 (CE); 5-HT promotes BDNF expression and TrkB activation, while BDNF affects 5-HTT function (DE). F. MGB disturbance is characterized by disturbed microbiota composition/metabolism. MGB-OS/mitochondria interaction (AF, F-OS): Microbiota affects ROS generation by modulating activities of NOX, SCFAs and antioxidants, and mitochondria metabolism; Mitochondrial genotype/mutation affects microbiota through mtROS. Neuroinflammation-MGB interaction (BF): MGB disturbance leads to gut inflammation, increases QUIN that damages BBB and results in neuroinflammation; Neuroinflammation disturbs microbiota homeostasis, through pro-inflammatory cytokines and NLRP3. BDNF-MGB interaction (DF): BDNF modulates intestinal barrier integrity; MGB disturbance reduces hippocampal BDNF expression. 5-HT-MGB interaction (EF): Microbiota disturbance inhibits 5-HT generation, by promoting intestinal/central KP, affecting 5-HT and TPH 1 gene expressions; 5-HTT genotype modulates microbiota composition. G. HPA axis interacts with multiple aspects, whose dysregulation attenuates the influence correspondingly: HPA affects ROS generation, mitochondria gene expression and metabolism through GRs, while NO mediates IL-1β and cholinergic stimulation of HPA (G-OS); HPA interacts with neuroinflammation through cytokines and GR/NF-κB signaling (BG); HPA affects glutamate release (through GR-TrkB interaction) and related gene expression (CG); GR activation reduces BDNF expression, while BDNF affects GR phosphorylation and rewrote glucocorticoid transcriptome (DG); HPA hormones increase intestinal barrier permeability and disturb microbiota composition, while MGB affects HPA through GABAergic activity (FG). AB, BC, CD, DE, EF and AF have indirect interaction through OS. AP-1 = activating protein-1; AMPA = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic; ATP = adenosine triphosphate; BDNF = brain-derived neurotrophic factor; CREB = cAMP responsive element binding protein; EAATs = excitatory amino acid transporters; eEF2 = eukaryotic elongation factor 2; ERK = extracellular signal-related kinases; GS = glutamine synthetase; GLS = glutaminase; Glu = glutamate; GLT-1 = glutamate transporter 1; IDO = indoleamine 2,3-dioxygenase; IL-1β = interleukin-1β; IL-6 = interleukin-6; KMO = kynurenine 3-monooxygenase; KP = kynurenine pathway; MGB = microbial-gut-brain; mPFC = medial prefrontal cortex; mtROS = mitochondrial ROS; NF-κB = nuclear factor kappa-B; NLRP3 = NOD-like receptor thermal protein domain associated protein 3; NMDA = N-methyl-D-aspartic acid; NOX = NADPH oxidase; OS = oxidative stress; OXPHOS = oxidative phosphorylation; PLC-γ = phospholipase C-γ; PKC = protein kinase C; PI3K = phosphatidylinositide 3-kinase; PVN = paraventricular nucleus; QUIN = quinolinic acid; RNS = reactive nitrogen species; ROS = reactive oxygen species; SCFAs = short-chain fatty acids; SIRT3 = sirtuin 3; SRC =sparse representation-based classification; TDO =tryptophan-2,3-dioxygenase; TPH = tryptophan hydroxylase; TNF-α = tumor necrosis factor-α; TrkB = tyrosine kinase B; TRYCATs = tryptophan catabolites; VGLUT1 = vesicular glutamate transporter 1; 5-HTT = serotonin transporter.