Figures & data
Figure 1 Pathogenesis of alcohol-related HCC. Alcohol undergoes metabolism in the liver to acetaldehyde, inducing the production of CYP2E1. Acetaldehyde, a carcinogenic substance, forms adducts with DNA interstrand cross-links. It diminishes the activity of O6-methylguanine DNA methyltransferase, resulting in insufficient DNA repair and, collectively, leading to DNA mutation. Alcohol disrupts gut microbial homeostasis, producing PAMPs and DAMPs. This process activates hepatocytes, Kupffer cells, and hepatic stellate cells (HSCs), leading to the secretion of chemokines, inflammation, and induced production of ROS. The ROS-produced malondialdehyde (MDA) forms adducts with acetaldehyde, resulting in DNA mutations. Alcohol induces ectopic upregulation of TLR4, which recognizes two patterns, releasing pro-inflammatory IL-8 and IL-1β. Moreover, the TLR4 downstream gene Nanog mediates TLR4-dependent hepatocellular carcinogenesis via hepatic progenitor cells. Alcohol also promotes the formation of HCC by down-regulating ferredoxin levels and inhibiting CD8+ cells and transcription of miRNA-122 and miRNA-483. (→, promote; ⊣, inhibit).
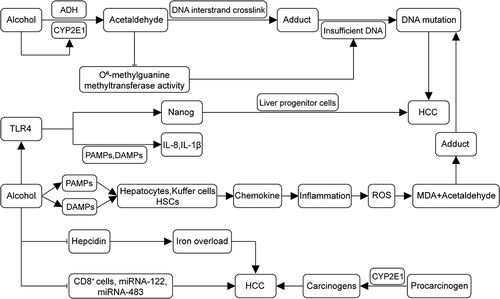
Figure 2 Mechanisms of insulin resistance and lipid accumulation in the development of MASLD-associated HCC. IGF-1 synthesis is enhanced in MASLD, with IGF-1 binding to IGF1R and activating IRS-1. IRS-1 further triggers MAPK, leading to the upregulation of proto-oncogenes (eg, c-fos and c-jun). MAPK activates the Wnt/β-catenin pathway, promoting tumor formation. IRS-1 also activates the PI3K/AKT pathway, contributing to tumorigenic effects. Insulin resistance upregulates GHR expression, binding to GH and promoting IGF-1 synthesis, activating the pro-cancer pathway. In cancer cells, GH binds to GHR to activate JAK2, accelerating the activation of TAT3, IRS-1, AKT, and ERK. Lipid accumulation increases IL-6, TNF-α, and leptin levels while decreasing lipocalin levels. IL-6 activates the JAK/STAT pathway, inducing HCC cell proliferation and tumor formation. TNF-α induced IL-6, regulating cancer cell differentiation, metastasis, and angiogenesis. TNF-α and IL-6 activate AP-1, NF-κ B, and STAT3 oncogenic transcription factors. Lipocalin activates AMPK signaling, inhibiting TNF-α expression, thereby increasing the risk of developing HCC. Leptin interacts with PI3K, MAPK, and JAK2/STAT signaling pathways, hastening HCC progression. (→, promote; ⊣, inhibit).
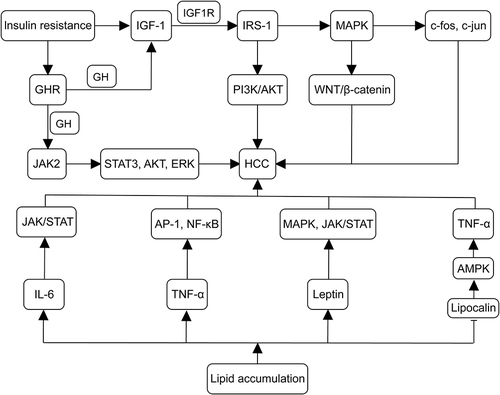
Figure 3 Mechanisms of miRNAs in the development of MASLD-associated HCC. miRNA-21 plays a role in tumor metabolism and activation during the progression of MASLD to HCC through PI3K/AKT, TGF-β and STAT3 signaling. The miR-34a-CDK6 signaling axis promotes MASLD development in high-fat environments. Deletion of the miRNA-122a increases the incidence of spontaneous HCC. The abundance of miRNA-301a-3p, miRNA-34a-5p, and miRNA-375 correlates with the progression of MASLD to HCC. Liver-specific expression of miR-378 inhibits FAO and promotes hepatic steatosis. miR-21-5p regulates PPARα expression, which decreases the oxidation of fatty acids, promoting lipotoxic lipid accumulation, and induces cell cycle activation, proliferation, and tumorigenesis. (→, promote; ⊣, inhibit).
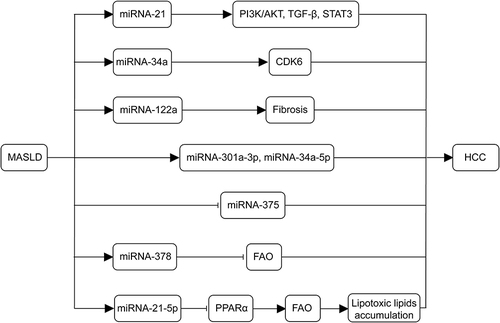
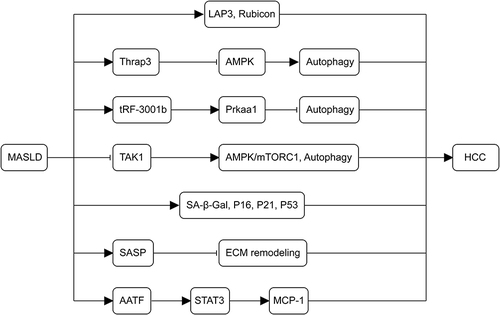
Figure 4 Mechanisms of autophagy and senescence in the development of MASLD-associated HCC. The upregulation of LAP3, Thrap3, tRF-3001b, and the autophagy-inhibitory protein Rubicon in MASLD inhibits autophagy. The deletion of TAK1 leads to chronic inflammation, fibrosis, and carcinogenesis in the liver by blocking the AMPK/mTORC1 pathway and autophagy. As MASLD advances, senescence-related markers (SA-β-Gal, p16, p21, and p53) persistently increase, contributing to the pathogenesis of MASLD-HCC. SASP worsens cellular senescence through autocrine and paracrine secretion, impedes ECM remodeling, and promotes tumor formation. AATF, overexpressed in MASLD, MASH, and MASLD-HCC via STAT3, enhances MCP-1 expression, promoting HCC.