
Figures & data
Figure 1 Functions of ROS at physiological level. Besides their direct functions in immune defense and cellular structure, ROS can act as second messengers. They oxidize the three main sulfur switches and indirectly influence pro-fibrotic signaling, autophagy, mitoptosis, pro-inflammatory signaling, apoptosis and cell proliferation. These processes are mediated by kinases, phosphatases and transcription factors, among others.
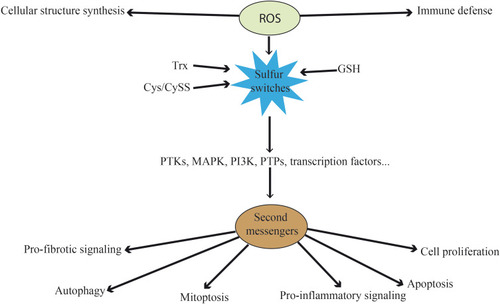
Figure 2 Inflammation and ROS. Elevated ROS production as a result of inflammatory signaling can mediate canonical NF-κB activation and downstream inflammatory gene induction, proteasome activity, antioxidant gene transcription, inflammasome activation, and cytokine secretion. The intricate balance between cell death and cell survival is largely modulated by intracellular ROS generation. In general, high intracellular ROS generation causes cell death by activation of cell death pathways (mitochondrial-dependent and -independent), whereas low levels of ROS act as signaling molecules that facilitate cell survival. Death receptors such as TNFR1 cause cell death by inducing high intracellular ROS production. The binding of TNF-α to TNFR1 recruits proteins such as TRADD and TRAF2 to activate TNFR1 signaling. The associated TRADD further recruits FADD and pro-caspase-8, and this whole signaling complex known as DISC is endocytosed, resulting in caspase-8 activation. Similar to TNFR1, the extent of caspase-8 activation determines whether a cell will follow a mitochondrial-dependent pathway or an independent pathway. Low caspase-8 activation follows a mitochondrial-dependent amplification loop which causes Cyt-C release. The released Cyt-C then subsequently binds Apaf-1 and caspase-9 and activates effector caspases such as caspase-3, which causes cell death. In the event of significant caspase-8 activation, it directly activates caspase-3 independent of mitochondria. High intracellular ROS induce sustained JNK activation and causes mitochondrial Cyt-C-dependent cell death. TNFR1 signaling also activates the NF-κB transcription factor by recruiting TRAF-2 and RIPK1 proteins. NF-κB activation is involved in the transcription of anti-apoptotic signaling proteins such as cIAP-2 and Bcl-xL. On the other hand, TNF can activate the NOX1 (neutrophilic) or NOX2 (epithelial) complex. The first step of NOX1/2 activation is the interaction of the cytosolic domain of TNFR1 with RFK and p22phox. The activated NOX complex converts extracellular O2 into O2−•, which extracellular SOD3 then converts into extracellular H2O2. The generated H2O2 passes freely through the plasma membrane and acts as a major source of intracellular ROS. Furthermore, TNF-induced formation of complex II leads to interactions between activated caspase-8, ROMO1 and Bcl-XL in the outer mitochondrial membrane, which reduce mitochondrial membrane potential, triggering MOMP and the production of MtROS. Conversely, if the levels of caspase-8 are high, TNF can activate directly caspase-3 independent of mitochondria, leading to apoptosis. Additionally, Ca2+-activated DUOX produces even more H2O2, which joins the cycle. Another important role of MtROS is the regulation of the inflammasome. MtROS are involved in immune cell activation during inflammation and are primary regulators of NLRP3 inflammasome activation. These high-molecular-weight complexes activate inflammatory caspases (caspase-1 and −11) and cytokines (IL-1β and IL-18). Also illustrated are ROS activating NF-κB directly or indirectly by inhibiting IKK phosphatases, and JNK stimulating mitochondrial ROS production through SAB.
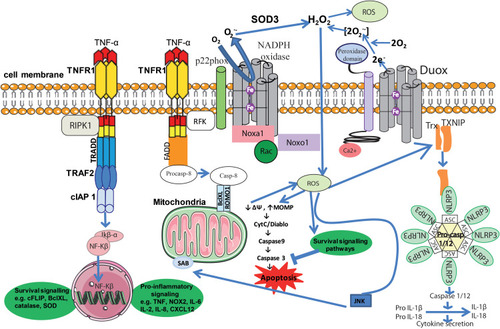
Figure 3 Role of ROS in neutrophil migration through endothelial cells. Integrins bind to ICAM-1 expressed in activated endothelial cells and arrest neutrophils on the endothelial surface. This leads to the activation of Cdc42 and Rac, which induce intracellular ROS generation through NOX. Increased ROS are involved in activation of different kinases, including c-Src and PKC, and thus induce phosphorylation and destabilization of endothelial junction proteins such as adherens junction. Moreover, ROS enhance the expression of P-selectin on the endothelium. Furthermore, ROS are known to activate NF-κB, inducing the expression of cell adhesion molecules such as ICAM-1, VCAM-1 and E-selectin which, in turn, enhance neutrophil binding on the endothelium. Acute exposure to pro-inflammatory factors triggers NOX-mediated ROS, which results in an increase of [Ca2+] by TRPC/TRPM ion channels. Increased [Ca2+] activates calmodulin and several downstream kinases including myosin light chain kinase (MLCK), which contribute to the disassembly of endothelial junction proteins.
![Figure 3 Role of ROS in neutrophil migration through endothelial cells. Integrins bind to ICAM-1 expressed in activated endothelial cells and arrest neutrophils on the endothelial surface. This leads to the activation of Cdc42 and Rac, which induce intracellular ROS generation through NOX. Increased ROS are involved in activation of different kinases, including c-Src and PKC, and thus induce phosphorylation and destabilization of endothelial junction proteins such as adherens junction. Moreover, ROS enhance the expression of P-selectin on the endothelium. Furthermore, ROS are known to activate NF-κB, inducing the expression of cell adhesion molecules such as ICAM-1, VCAM-1 and E-selectin which, in turn, enhance neutrophil binding on the endothelium. Acute exposure to pro-inflammatory factors triggers NOX-mediated ROS, which results in an increase of [Ca2+] by TRPC/TRPM ion channels. Increased [Ca2+] activates calmodulin and several downstream kinases including myosin light chain kinase (MLCK), which contribute to the disassembly of endothelial junction proteins.](/cms/asset/94aed9fd-65a7-4958-8b98-681077ae8b46/djir_a_12198534_f0003_c.jpg)