Figures & data
Figure 1. Wip1 protein abundance during the cell cycle. (A) HeLa cells were synchronized by a double thymidine block, released into fresh media supplemented or not with nocodazole, and samples were collected at 2-h intervals and probed with indicated antibodies. pSer10-H3 was used as a marker of mitotic entry; degradation of cyclin A as a marker of prometaphase and degradation of cyclin B as a marker of mitotic exit. (B) Asynchronously growing FUCCI indicator expressing U2OS cells were pretreated with Hoechst DNA dye and the following populations of cells were sorted: double-negative (DN) and single RFP-positive cells (RNF+); single GFP-positive cells (GFP+); double-positive (DP) cells and samples were analyzed by flow cytometry. Note that the DN/RFP+ population corresponds to cells with a low DNA content (G1 phase), whereas GFP+ population corresponds to 4 n cells (G2 phase) and DP show intermediate DNA content (S phase). (C) Populations of cells from (B) analyzed by immunoblotting. Cyclin D was used as a marker of G1, cyclin A and Plk1 as markers of G2. (D) U2OS and RPE cells were cotransfected by Wip1 shRNA plasmid (shWip1) together with a mCherry marker and probed with polyclonal Wip1 (sc20712) or monoclonal Wip1 (sc37625) antibodies and with DAPI. Neighboring untransfected cells were used as a control (mock). Shown is quantification of immunofluorescence staining in interphase and mitotic cells. Note higher Wip1 signal intensity in cells with higher DAPI corresponding to G2 cells and decreased Wip1 signal in mitotic cells. As additional control of signal specificity, cells were treated with Etoposide (2.5 µM for 16 h), which caused accumulation of G2 cells with high intensity of Wip1. (E) Asynchronously growing U2OS cells were fixed and probed with indicated antibodies and DAPI. Note decreased Wip1 signal in mitosis and absence of the signal on mitotic chromatin.
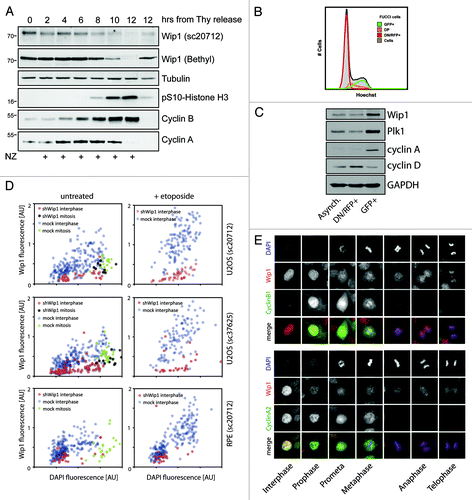
Figure 2. Degradation of Wip1 during mitosis. (A) U2OS cells were synchronized by a double thymidine block, released into media with NZ and after 8 h, treated or not with 5 μM MG132. (B) Asynchronously growing cells were treated for 60 min with 5 µM RO-3306 to release cells from mitosis and prevent new cells from entering. Subsequently, cells were treated for 60 min with MG132 (5 μM) or DMSO, fixed, stained for Wip1 and the signal was quantified. Each dot corresponds to one cell. The dashed lines indicate the respective mean values. The difference between the means was analyzed by Student’s t-test (** p < 0.01). (C) Cells were synchronized in late G2 by Ro-3306 and released into fresh media with NZ for 2 h. Mitotic cells were collected by shake-off and analyzed by immunoblotting (left) or qPCR (right). (D) Cells were transfected with siRNAs targeting indicated proteins. One-third of the cells were synchronized for last 24 h with thymidine, and adherent cells were collected. One-third of the cells were treated with NZ for the last 12 h and collected by mitotic shake-off. Cell extracts were collected at 48 h after transfection and probed with indicated antibodies. RNA was extracted from the last part of cells and analyzed by qPCR (Fig. S3). (E) Cells were transfected with siRNAs targeting cdc20 or GAPDH (negative control). Mitotic cells were collected by shake-off 24 h after transfection and probed with indicated antibodies.
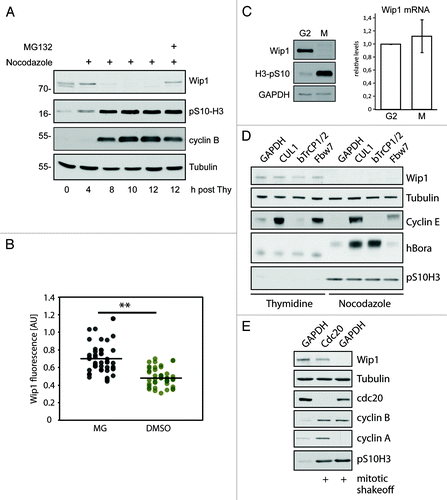
Figure 3. Wip1 is extensively phosphorylated in mitosis. (A) Expression of Wip1-FLAG was induced by tetracycline and Wip1-FLAG was immunoprecipitated from asynchronously growing cells (Asynch.), from mitotic cells arrested in NZ and collected by shake-off (Mitotic) or from cells treated with 0.5 μM adriamycine (Damage). Half of the immunopurified material was subjected to treatment with lambda phosphatase. (B) Cells expressing Wip1-D314A-FLAG were arrested in G2 by treatment with adriamycine and forced to enter mitosis by treatment with caffeine. Alternatively, roscovitine was added to prevent mitotic entry. (C) Cells expressing Wip1-D314A-FLAG were arrested in mitosis by treatment with NZ and released into fresh media for indicated time. Alternatively, mitotic exit was blocked by addition of MG132. (D) Cells expressing Wip1-FLAG were arrested in mitosis by NZ and treated with DMSO, roscovitine (12 μM) or p38i (3 μM, SB202190) for 90 min. Cell lysates were separated on Phostag SDS-PAGE and probed with indicated antibodies. (E) Phosphatase dead His-Wip1-D314A was subjected to in vitro kinase assay with Cdk1/cyclin B, separated on SDS-PAGE and phosphorylation was detected by autoradiography. (F) Wip1-D314A-FLAG was purified from mitotic cells and analyzed by MS. Scheme of identified phosphorylated residues is shown. (G) His-Wip1 or His-Wip1-S40A was subjected to in vitro kinase assay with Cdk1/cyclin B, separated on SDS-PAGE and phosphorylation was detected by autoradiography. (H) Cells expressing Wip1-FLAG, Wip1-S40A, Wip1-S46A or Wip1-AAA were synchronized in mitosis by NZ or grown asynchronously. Note a partial loss of mobility shift in the S40A mutant purified from mitotic cells. (I) Cells expressing Wip1-WT or Wip1–7A were grown asynchronously or arrested in mitosis by addition of nocodazole for 12 h and probed with indicated antibodies. Note decreased mobility shift of Wip1–7A in mitotic cells.
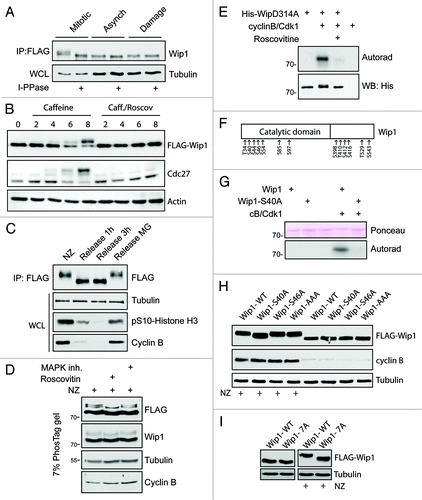
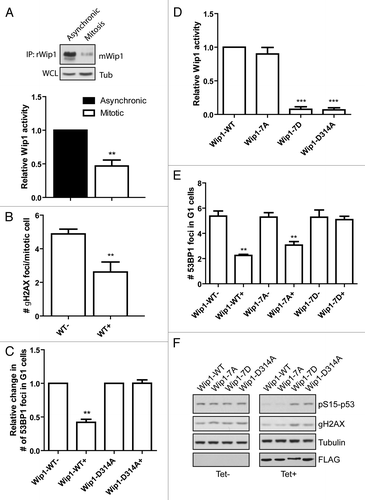
Figure 4. Activity of Wip1 is decreased in mitosis. (A) Endogenous Wip1 was immunoprecipitated using rabbit anti-Wip1 (H300) from asynchronously growing or NZ-arrested cells and phosphatase activity was measured in vitro. (B) Expression of Wip1-WT was induced or not by tetracycline in asynchronously growing cells. Number of γH2AX positive foci was determined by a manual evaluation of immunofluorescence staining in 70 mitotic cells (n = 3, error bars indicate ,, ** p < 0.01). (C) Expression of Wip1 or Wip1-D314A was induced (+) or not (-) by tetracycline and cells were probed for 53BP1. Number of 53BP1 positive nuclear bodies in G1 cells was determined by automated microscopy in 1000 cells (n = 3, error bars indicate SD, ** p < 0.01). (D) Expression of Wip1-WT, Wip1–7A, Wip1–7D and Wip1-D314A was induced by tetracycline, FLAG-tagged proteins were immunoprecipitated and the phosphatase activity was determined in vitro (n = 3, error bars indicate SD, ** p < 0.01). (E) Expression of Wip1-WT, Wip1–7A, Wip1–7D and Wip1-D314A was induced (+) or not (-) by tetracycline and number of 53BP1 nuclear bodies was determined by automated microscopy. Intensity of DAPI signal was used for gating of G1 cells (n = 3, error bars indicate SD, ** p < 0.01). (F) Expression of Wip1-WT, Wip1–7A, Wip1–7D and Wip1-D314A was induced (+) or not (-) by tetracycline; cells were treated with 0.5 μM adriamycine for 4 h, and whole-cell lysates were probed for pSer15p53 and γH2AX.