Figures & data
Figure 1. Ethanol induces Cav-1 downregulation and myofibroblastic differentiation in the stromal fibroblast compartment. (A) Fibroblasts were cultured for 2 d. To study the effect of ethanol (EtOH), cells were cultured in the presence or in the absence of 100 mM EtOH for additional 72 h. Western blot analysis using antibody directed against Cav-1. Note that EtOH exposure induces a downregulation of Cav-1 in fibroblasts. β-actin was used as equal loading control. (B) Immunofluorescence. MCF7 cells were co-cultured with fibroblasts for 2 d and treated with 100 mM EtOH for additional 72 h. Then, cells were fixed and immunostained with antibody probes against Cav-1 (red, upper panels) and keratin-8/18 (K8/18, green, lower panels). Nuclei were counterstained with DAPI (blue). K8/18 staining is specific for MCF7 cells and Cav-1 is specific for fibroblasts, since MCF7 cells lack Cav-1 expression. Note that Cav-1 expression is downregulated in the fibroblast compartment after ethanol treatment, relative to untreated cells. Original magnification, 40x. (C) Fibroblasts were allowed to growth for 2 d, and then were cultured in the presence or in the absence of 100 mM EtOH for 72 h. Western blot analysis using antibodies against vimentin and SMA. β-actin was used as equal loading control. Note that ethanol-treated fibroblasts display an upregulation of the myofibroblast markers SMA and vimentin.
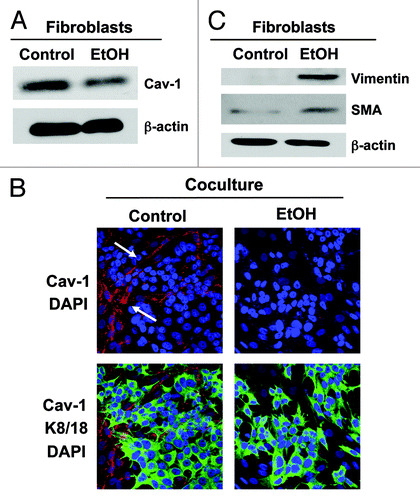
Figure 2. Ethanol treatment drives oxidative stress in fibroblasts. Fibroblasts were plated in homotypic culture or in co-culture with RFP(+)-MCF7 cells. After 2 d, cells were treated with 100 mM EtOH for 72 h. Untreated cells served as controls. ROS production was determined by flow cytometry using CellROX Deep Red Reagent probe. Fibroblasts were identified as the RFP(-)-cell population. Note that in fibroblasts cultured alone, EtOH treatment increases ROS production by 35%, as compared with control cells. When fibroblasts were co-cultured with cancer cells, the increase in ROS production is even higher, rising up to 58%. p values are as indicated.
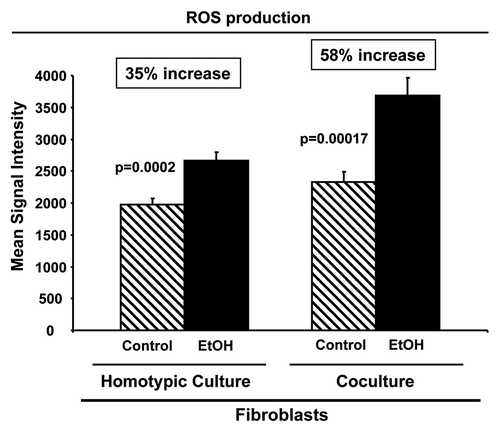
Figure 3. Ethanol induces autophagy in fibroblasts, but promotes autophagy resistance in MCF7 cancer cells. (A and B) Fibroblasts were cultured in the presence or in the absence of 100 mM EtOH for 72 h. (A) Western blot analysis using antibodies against a panel of autophagy markers. β-actin was used as equal loading control. Note that EtOH treatment increases the expression of several key autophagy markers, namely BNIP3, LC3, Cathepsin B and, to a lesser extent, Lamp-1. LC3 activation is also increased, as detected by cleavage (LC3-II). (B) Western blot analysis using antibodies against phospho-AKT. The membranes were then reprobed with total AKT antibodies. Note that ethanol treatment promotes activation of the AKT pathway. This reaction is probably an anti-apoptotic response to protect the fibroblasts against autophagy-induced cell death. (C) Immunofluorescence. MCF7 cell-fibroblast co-cultures were maintained in the presence or absence of 100 mM EtOH for 72 h. Cells were fixed and immunostained with antibody probes directed against K8/18 (green, to identify the MCF7 cell population), and Cathepsin B, LC3 or BNIP3 (all red, to detect autophagy). Nuclei were counterstained with DAPI (blue). To detect LC3 prior to fixation, cells were incubated with HBSS in the presence of 25 µM chloroquine for 6 h. The upper panels show the control, untreated conditions. The bottom panels show the ethanol-treated samples. Note that autophagy markers (Cathepsin B, LC3 and BNIP3) were all selectively upregulated in the fibroblast compartment in response to ethanol treatment. Original magnification, 40x, except for LC3, 63x. (D) MCF7 cells were cultured in the presence or in the absence of 100 mM EtOH for 72 h. Western blot analysis using antibodies against a panel of autophagy markers. β-actin was used as equal loading control. Note that EtOH treatment decreases the expression of several key autophagy markers, namely BNIP3, LC3-I (precursor form), LC3-II (active cleaved form), Cathepsin B and, to a lesser extent, Lamp-1, suggesting that ethanol promotes autophagy resistance in MCF7 cancer cells.
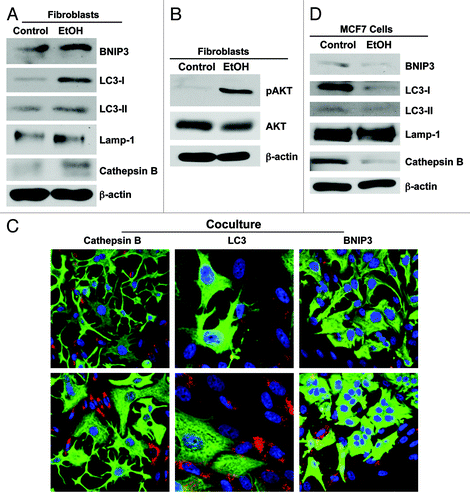
Figure 4. Ethanol increases the expression of MCT4 in cancer-associated fibroblasts. MCF7 cells were plated in co-culture with fibroblasts, and cells were treated with 100 mM EtOH for 72 h. Cells were fixed and immunostained with antibodies against MCT4 (red, upper panels) and K8/18 (green, lower panels). Nuclei were counterstained with DAPI (blue). Note that treatment with EtOH greatly increases the expression of MCT4 in cancer-associated fibroblasts. Original magnification, 40x.
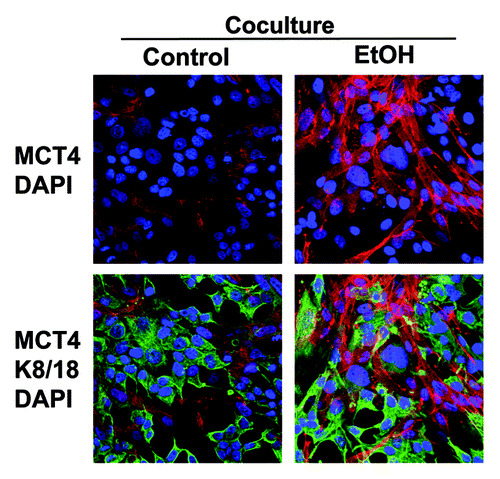
Figure 5. Ethanol promotes ketone body generation in fibroblasts. (A) β-hydroxybutyrate (β-OH-butyrate) concentration was measured in the cell culture media derived from fibroblasts cultured in the presence or absence of 100 mM EtOH for 72 h. Values were normalized either for protein concentration (left) or cell number (right). Note that EtOH treated-fibroblasts display a 1.5-fold increase in β-OH-butyrate accumulation, relative to untreated fibroblasts. (B) Western blot analysis. Control and EtOH-treated fibroblasts were analyzed by immunoblotting with a panel of antibodies against key enzymes of ketone body production. β-actin was used as equal loading control. Note that treatment with EtOH induces the expression of the enzymes involved in ketone production (HMGCS1, HMGCS2 HMGCL and BDH1). These data indicate that ethanol increases ketone body generation in fibroblasts.
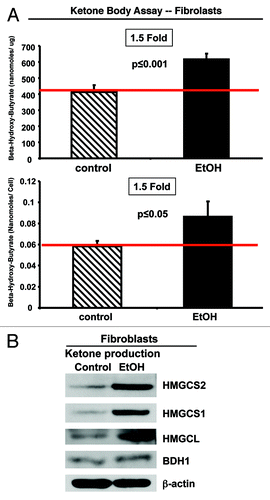
Figure 6. Ethanol promotes ketone body utilization in MCF7 cells. (A) Western blot analysis. Control and EtOH-treated MCF7 cells were analyzed by immunoblotting with a panel of antibodies against key enzymes of ketone body metabolism (production and utilization). β-actin was used as equal loading control. Note that treatment with EtOH induces the expression of key enzymes involved in ketone utilization (ACAT-1 and OXCT1). The expression of key enzymes involved in ketone body production (HMGCS1, HMGCS2 and HMGCL) is unchanged or slightly decreased. (B and C) Immunofluorescence. Fibroblast-MCF7 cell co-cultures were treated with 100 mM EtOH for 72 h. Cells were then fixed and immunostained with antibodies against K8/18 (green) and ACAT-1 [red, (B) or OXCT-1 red, (C)]. Nuclei were counterstained with DAPI (blue). The upper panels show ACAT-1 or OXCT-1 staining (red). The bottom panels show also the K8/18 staining (green) to identify the MCF7 cell population. Note that ethanol increases the expression of enzymes for ketone bodies utilization [ACAT-1, (B); and OXCT-1, (C)] in cancer cells in co-culture with fibroblasts. Original magnification, 40x. These data indicate that ethanol increases ketone body utilization in MCF7 cancer cells.
![Figure 6. Ethanol promotes ketone body utilization in MCF7 cells. (A) Western blot analysis. Control and EtOH-treated MCF7 cells were analyzed by immunoblotting with a panel of antibodies against key enzymes of ketone body metabolism (production and utilization). β-actin was used as equal loading control. Note that treatment with EtOH induces the expression of key enzymes involved in ketone utilization (ACAT-1 and OXCT1). The expression of key enzymes involved in ketone body production (HMGCS1, HMGCS2 and HMGCL) is unchanged or slightly decreased. (B and C) Immunofluorescence. Fibroblast-MCF7 cell co-cultures were treated with 100 mM EtOH for 72 h. Cells were then fixed and immunostained with antibodies against K8/18 (green) and ACAT-1 [red, (B) or OXCT-1 red, (C)]. Nuclei were counterstained with DAPI (blue). The upper panels show ACAT-1 or OXCT-1 staining (red). The bottom panels show also the K8/18 staining (green) to identify the MCF7 cell population. Note that ethanol increases the expression of enzymes for ketone bodies utilization [ACAT-1, (B); and OXCT-1, (C)] in cancer cells in co-culture with fibroblasts. Original magnification, 40x. These data indicate that ethanol increases ketone body utilization in MCF7 cancer cells.](/cms/asset/5e8fc3f6-9de6-425e-a60a-581b00ca2997/kccy_a_10923109_f0006.gif)
Figure 7. Ethanol increases the mitochondrial activity in MCF7 cancer cells. (A) MCF7 cells were cultured in the presence or in the absence of 100 mM EtOH for 72 h. Western blot analysis using antibodies against subunits of the mitochondrial OXPHOS respiratory chain (complexes I–V). β-actin is shown as equal loading control. Note that ethanol significantly increases the expression of OXPHOS complex I (20 kDa subunit) and IV (COX-II) in MCF7 cancer cells. (B) Immunocytochemistry. MCF7 cell-fibroblast co-cultures were treated with 100 mM EtOH for 72 h. Cells were fixed and immunostained with antibodies against the mitochondrial membrane marker TOMM20 and K8/18. Nuclei were counterstained with DAPI (blue). The upper panels show the mitochondrial staining (red). The bottom panels show also the K8/18 staining (green), to identify the MCF7 cell population. Note that ethanol increases the mitochondrial mass specifically in MCF7 cells, as compared with untreated cells. However, ethanol does not promote mitochondrial biogenesis in fibroblasts. Original magnification, 40x.
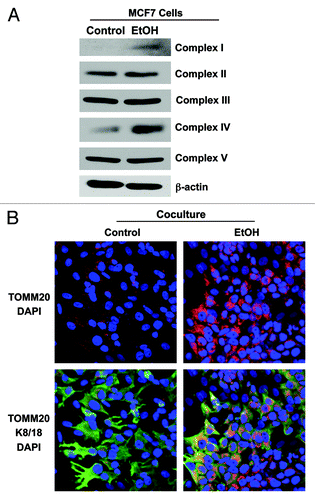
Figure 8. Ethanol increases the amounts of ATP-rich vesicles in MCF7 cancer cells. RFP(+)-MCF7 cells were plated in homotypic culture or in co-culture with fibroblasts. Cells were treated with 100 mM EtOH for 72 h. To detect ATP-rich vesicles, cells were incubated with 30 µM quinacrine in phenol-red-free DMEM for 1 h at 37°C. The green fluorescence of ATP-filled vesicles was recorded by flow cytometry. Note that ethanol treatment promotes a 45% increase in ATP-enriched vesicles when cancer cells are co-cultured with fibroblasts, without affecting quinacrine uptake of MCF7 cells cultured alone. p values are as indicated.
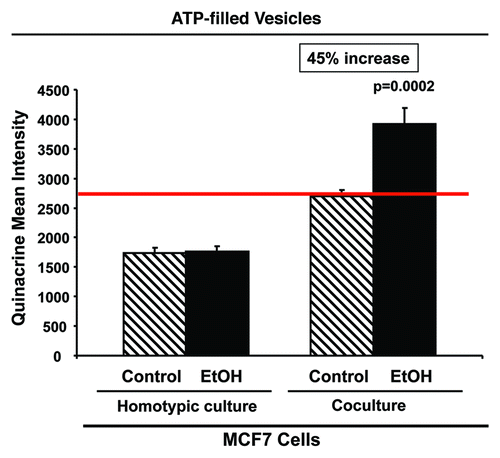
Figure 9. Ethanol suppresses the expression of ERα in MCF7 cancer cells. MCF7 cell-fibroblast co-cultures were treated with 100 mM EtOH for 72 h. Cells were fixed and immunostained with antibody probes directed against ERα (red) and K8/18 (green). Nuclei were counterstained with DAPI (blue). The upper panels show ERα staining only (red). The bottom panels show also the K8/18 staining (green) to identify the MCF7 cell population. Note that ethanol suppresses the expression of ERα specifically in MCF7 cells, as compared with untreated cells. Original magnification, 40x.
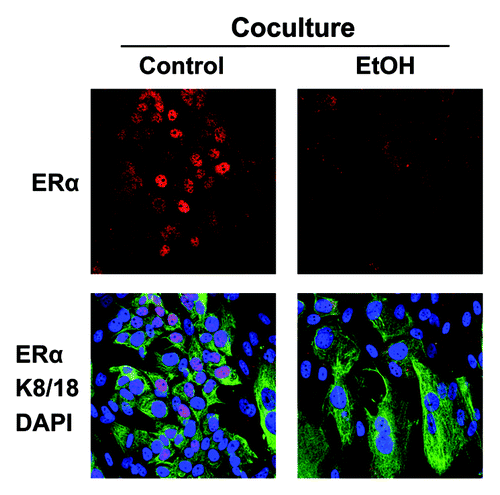
Figure 10. Ethanol does not promote apoptotic cell death. RFP(+)-MCF7 cells were plated in homotypic culture or in co-culture with fibroblasts. Cells were treated with 100 mM EtOH for 72 h. Apoptosis was detected with annexin V-APC and PI by flow cytometry. Note that MCF7 cells are protected against cell death when co-cultured with fibroblasts (A). However, ethanol does not significantly promote apoptosis in MCT7 cells (A) or in fibroblasts (B).
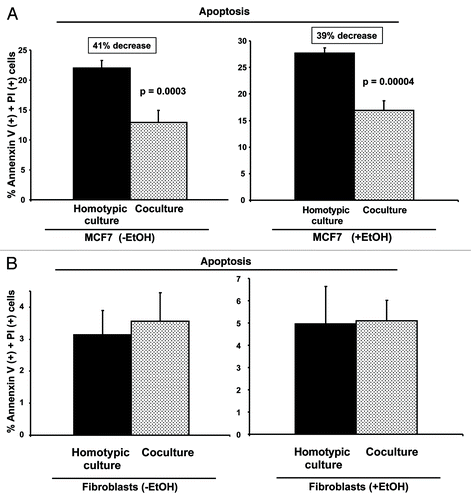
Figure 11. Anti-oxidant treatment prevents the ethanol-induced loss of Cav-1 in cancer-associated fibroblasts. MCF7 cells were co-cultured with fibroblasts for 2 d, and treated with 100 mM EtOH for an additional 72 h. Then, 10 mM NAC or vehicle was added for 24 h. The cells were fixed and immunostained with antibody probes against Cav-1 (red, upper panels) and keratin-8/18 (K8/18, green, lower panels). Nuclei were counterstained with DAPI (blue). K8/18 staining is specific for MCF7 cells and Cav-1 is specific for fibroblasts, since MCF7 cells lack Cav-1 expression. Note that treatment with the antioxidant NAC prevents the downregulation of Cav-1 in the fibroblast compartment, as compared with co-cultured fibroblasts in the absence of NAC. Original magnification, 40x.
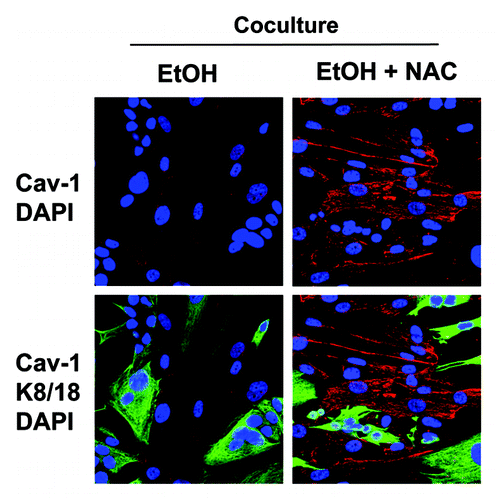
Figure 12. Antioxidant treatment prevents the ethanol-induced upregulation of MCT4 in cancer-associated fibroblasts. MCF7 cells were plated in co-culture with fibroblasts, and cells were treated with 100 mM EtOH for 72 h. Then, 10 mM NAC or vehicle was added for 24 h. Cells were fixed and immunostained with antibodies against MCT4 (red, upper panels) and K8/18 (green, lower panels). Nuclei were counterstained with DAPI (blue). Note that treatment with the antioxidant NAC prevents the upregulation of MCT4 in fibroblasts compartment, as compared with co-cultured fibroblasts in the absence of NAC. Original magnification, 40x.
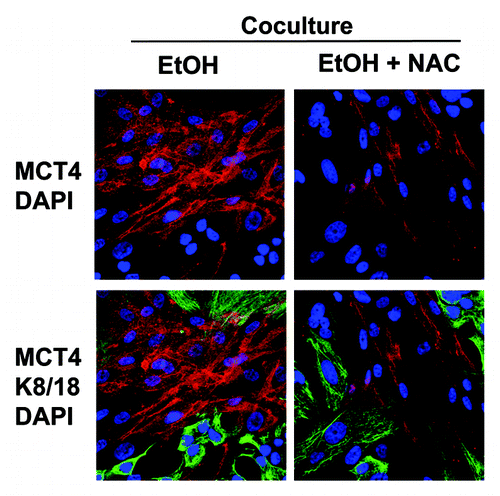
Figure 13. Alcohol consumption and two-compartment tumor metabolism. Ethanol is normally detoxified to acetyl-CoA, which functions as a mitochondrial fuel. However, the first metabolic intermediate is acetaldehyde, which is known to be a chemical carcinogen that results in DNA damage and increased ROS production. In fibroblasts, this initiates the onset of oxidative stress, driving a loss of Cav-1 (via autophagy) and promoting the myofibroblast phenotype [SMA(+)]. Ultimately, this leads to mitophagy and mitochondrial dysfunction in fibroblasts. In cells with dysfunctional mitochondria (fibroblasts), acetyl-CoA is converted to ketone bodies, which are then exported via MCT4 (monocarboxylate transporter 4). In contrast, in cells with functional mitochondrial (epithelial cancer cells), acetyl-CoA is burned in the TCA cycle via OXPHOS. Thus, fibroblasts convert ethanol to mitochondrial fuel (ethanol → acetyl-CoA → ketone bodies), which is exported to “feed” adjacent cancer cells. Then, cancer cells upregulate the necessary enzymes (ACAT/OXCT) to convert ketone bodies back into acetyl-CoA, fueling oxidative mitochondrial metabolism.
![Figure 13. Alcohol consumption and two-compartment tumor metabolism. Ethanol is normally detoxified to acetyl-CoA, which functions as a mitochondrial fuel. However, the first metabolic intermediate is acetaldehyde, which is known to be a chemical carcinogen that results in DNA damage and increased ROS production. In fibroblasts, this initiates the onset of oxidative stress, driving a loss of Cav-1 (via autophagy) and promoting the myofibroblast phenotype [SMA(+)]. Ultimately, this leads to mitophagy and mitochondrial dysfunction in fibroblasts. In cells with dysfunctional mitochondria (fibroblasts), acetyl-CoA is converted to ketone bodies, which are then exported via MCT4 (monocarboxylate transporter 4). In contrast, in cells with functional mitochondrial (epithelial cancer cells), acetyl-CoA is burned in the TCA cycle via OXPHOS. Thus, fibroblasts convert ethanol to mitochondrial fuel (ethanol → acetyl-CoA → ketone bodies), which is exported to “feed” adjacent cancer cells. Then, cancer cells upregulate the necessary enzymes (ACAT/OXCT) to convert ketone bodies back into acetyl-CoA, fueling oxidative mitochondrial metabolism.](/cms/asset/26064dba-43ae-4e67-8d3c-924cbede1b6c/kccy_a_10923109_f0013.gif)
Figure 14. Ethanol may increase breast cancer risk via metabolic reprogramming. In summary, we show that ethanol exposure dramatically affects the expression status of both stromal and epithelial biomarkers functionally associated with poor clinical outcome. For example, ethanol induces myofibroblastic differentiation [SMA(+)], autophagy [Cav-1(-)], as well as oxidative stress and mitochondrial dysfunction [MCT4(+)] in the tumor microenvironment, which fuels metabolic-coupling. Conversely, ethanol drives a shift toward ER(-) status, as well as mitochondrial biogenesis [TOMM20(+)] and ketone body re-utilization [ACAT1(+)/OXCT1(+)] as a mitochondrial energy source. Thus, ethanol “fertilizes” the microenvironment, leading to “two-compartment tumor metabolism” and field cancerization.
![Figure 14. Ethanol may increase breast cancer risk via metabolic reprogramming. In summary, we show that ethanol exposure dramatically affects the expression status of both stromal and epithelial biomarkers functionally associated with poor clinical outcome. For example, ethanol induces myofibroblastic differentiation [SMA(+)], autophagy [Cav-1(-)], as well as oxidative stress and mitochondrial dysfunction [MCT4(+)] in the tumor microenvironment, which fuels metabolic-coupling. Conversely, ethanol drives a shift toward ER(-) status, as well as mitochondrial biogenesis [TOMM20(+)] and ketone body re-utilization [ACAT1(+)/OXCT1(+)] as a mitochondrial energy source. Thus, ethanol “fertilizes” the microenvironment, leading to “two-compartment tumor metabolism” and field cancerization.](/cms/asset/536a1f06-a0e8-4cf6-91c7-874688d628bd/kccy_a_10923109_f0014.gif)