Figures & data
Figure 1A–D. Proteasome-dependent degradation of REV1. (A) Fast turnover of REV1 protein. HEK293T cells were transiently transfected with FLAG-REV1-expressing plasmid. Forty-eight hours after transfection, 15 μg/mL cycloheximide (CHX, lanes 5–8) or solvent (ethanol) alone (lanes 1–4) was added to cells, and cells were harvested at indicated time points after treatment. FLAG-REV1 levels were analyzed by western blotting with anti-FLAG antibody. Endogenous cyclin B1 (cyc B1) protein levels were also detected as a control for CHX activity. (B) FLAG-REV1 protein signals in A were quantified and plotted. Its half-lives (t1/2) upon addition of CHX and solvent were calculated as described.Citation74,Citation75 (C) REV1 is a polyubiquitinated protein. HEK293T cells were co-transfected with His-Ub and increasing amount of FLAG-REV1 expressing plasmid (lanes 1–2), or FLAG-REV1 and increasing amount of His-Ub (lanes 3–4). Cell lysates were pulled down with Ni-NTA resin in 6 M guanidine buffer followed by immunoblotting using anti-FLAG to detect ubiquitin-conjugated FLAG-REV1 (polyUb-REV1). One-fifth of harvested cells were lysed with RIPA to detect FLAG-REV1 (middle panel) and β-actin (lower panel). (D) REV1 is stabilized by proteasome inhibitor MG132. HEK293T cells were treated with increasing amount of proteasome inhibitor MG132 (10 and 20 μM) or DMSO. Cells were harvested at 0 h and 12 h after treatment and analyzed for endogenous REV1 protein expression with anti-REV1 antibodies. The membrane was also immunoblotted with anti-cyclin B1 antibody as a control for MG132 activity.
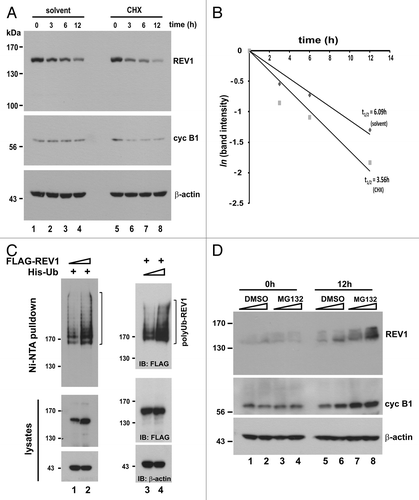
Figure 1E–F. (E) Cells were transiently transfected with same amount of FLAG-REV1 expression plasmid for 48 h. Increasing dose of MG132 (10 and 20 μM; lanes 5–6) or DMSO (lanes 2–3) was then added to cells. Cells were harvested 6 h after treatment and FLAG-REV1 was detected by western blotting with anti-FLAG. Relative levels of REV1 and cyclin B1 were determined by densitometry. (F) Inhibition of proteasome activity with MG132 attenuates REV1 degradation in HEK293T cells. Cells were transfected with FLAG-REV1 plasmid for 36 h. CHX was then added to cells (lanes 2–4). Meanwhile, cells were also supplemented with DMSO (lane 3) and 20 μM MG132 (lane 4), respectively. Cells were analyzed for FLAG-REV1 protein 6 h after drug treatment. Similar findings were also obtained from HeLa and IMR90 cells.
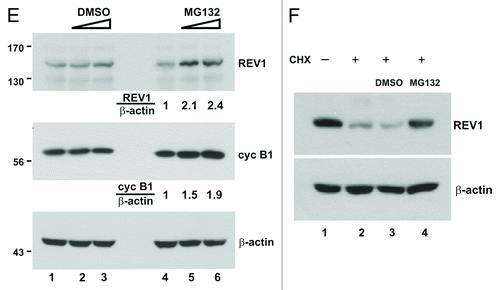
Figure 2. CDH1 and CDC20 promote REV1 degradation. (A) HEK293T cells were co-transfected with FLAG-REV1 plus empty vector (lane 1) or plus increasing dose of HA-CDH1-expression plasmids (lanes 2–3) for 48 h. FLAG-REV1 and HA-CDH1 proteins were detected with anti-FLAG and anti-HA antibodies, respectively. (B) Cells were co-transfected with FLAG-REV1 plus empty vector (lane 1) or plus increasing dose of Myc-CDC20 expression plasmids (lanes 2–3). (C) Proteasome inhibitor MG132 prevents CDH1-promoted degradation of REV1. Cells were transfected with FLAG-REV1 and HA-CDH1 (lanes 2–4) for 48 h. DMSO (lane 3) or 20 μM MG132 (lane 4) was added to cells for 6 h before harvest. (D) MG132 prevents CDC20-dependent degradation of REV1. Cells were co-transfected with FLAG-REV1 and HA-CDC20 plasmids and were then treated with MG132.
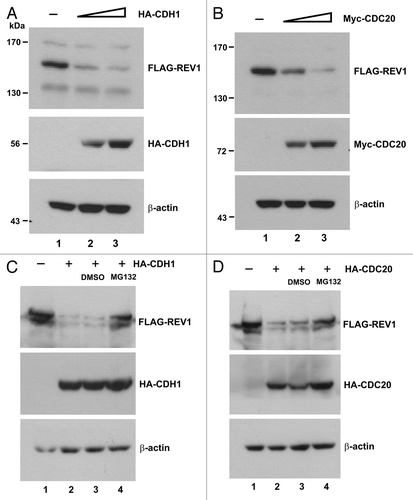
Figure 3. CDH1 and CDC20 enhance polyubiquitination of REV1. (A) Polyubiquitination of endogenous REV1. HEK293T cells were co-transfected with His-Ub plus empty HA (lane 1) or plus HA-CDH1 (lanes 2–3) expression plasmids. DMSO (lane 2) and MG132 (lane 3) were added to cells for 6 h before harvest. Cells were lysed in 6 M guanidine buffer and incubated with Ni-NTA beads. Ni-NTA pulldown assay was performed and polyubiquitinated REV1 was probed with anti-REV1 antibodies. RIPA lysed fractions were immunoblotted to detect HA-CDH1 and endogenous REV1 protein levels. ns, non-specific signal. Cells were co-transfected with FLAG-REV1 and His-Ub, plus increasing amount of HA-CDH1 (B) or Myc-CDC20 (C). Polyubiquitinated REV1 was detected by Ni-NTA pulldown assay and western blotting using anti-FLAG antibody. HA-CDH1 and Myc-CDC20 proteins in cell lysates were probed with anti-HA (B) and anti-MYC (C) antibodies, respectively. (D) REV1 interacts with anaphase APC component APC3. Cells were transfected with empty vector (lane 1) or FLAG-REV1 (lane 2), and the lysates were incubated with anti-FLAG antibody for immunoprecipitation. Precipitates were immunoblotted with anti-FLAG and anti-APC3 antibodies.
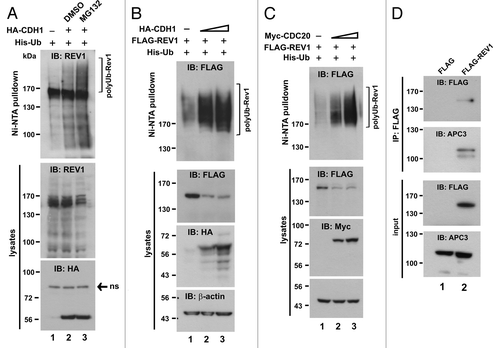
Figure 4. Requirement of the N-terminal part for REV1 degradation. (A) Schematic presentation of human REV1 protein (1–1,251 amino acids). D box, UBM1, UBM2, C-terminal REV7-binding domain (REV7 BD) and the two XbaI sites were indicated. Point mutations generated in D box, UBM1 and UBM2 were highlighted with an asterisk (*), and the change of the amino acid residues were specified. Truncated mutant ΔXbaI containing amino acids 1–885 of REV1 was generated by deleting the region between two XbaI sites. ΔBD is a REV1 mutant deleted of REV7 BD. All REV1 wild-type (wt) and mutant proteins were FLAG-tagged at their N termini. (B) Stability of REV1 wild-type and mutant proteins. HEK293T cells were transfected with FLAG-REV1 wild-type (wt) and mutants for 48 h. Cells were treated with 15 μg/mL CHX (lanes 2, 4, 6, 8 and 10) for 12 h and then harvested for western blotting using anti-FLAG antibody. (C) CDH1 promotes degradation of REV1 wild-type and mutant proteins. Cells were transfected with FLAG-REV1 wt and mutants as indicated, in the presence of HA-CDH1 (+) or empty HA-vector (−). FLAG-REV1 and HA-CDH1 proteins were detected by western blotting. (D) C-terminal REV7-binding domain is dispensable for REV1 degradation. Cells were transfected with increasing amounts of FLAG-REV1 ΔBD plasmid (1 μg in lanes 1, 3 and 5; 2 μg in lanes 2, 4 and 6) for 36 h. DMSO or proteasome inhibitor PSI was added to cells for 6 h before harvest. Similar results were also obtained with proteasome inhibitor MG132. (E and F) D-box, UBM and REV7 BD are dispensable for REV1 polyubiquitination. In (E), HEK293T cells were co-transfected with His-Ub plus FLAG-REV1 wt or plus ΔXbaI mutant for 48 h. Cell lysates were incubated with Ni-NTA beads in guanidine buffer to pull down polyubiquitinated REV1 protein, which was detected by western blotting. In (F), cells were co-transfected with FLAG-REV1 wt or mutants as indicated, and His-Ub, in the presence of HA-CDH1 (+) or HA-vector (−). Polyubiquitinated REV1 was pulled down by Ni-NTA beads and analyzed by western blotting using anti-FLAG antibody.
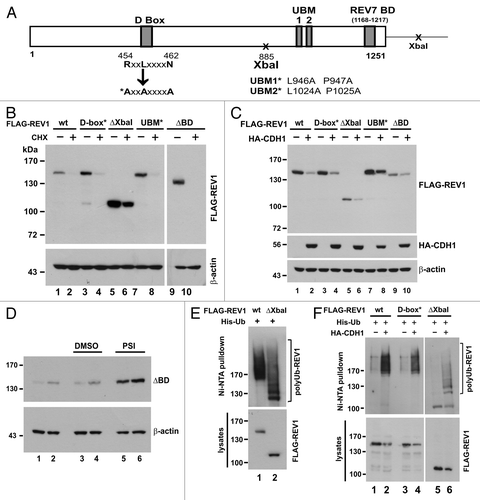
Figure 5. Interaction between REV1 and REV7. (A) Identification of a novel REV7-binding domain. HEK293T cells were co-transfected with FLAG-REV1 wt or mutants as indicated, and V5 His-REV7. FLAG-REV1 885–1,167 truncation mutant contains the C-terminal region (amino acids 885–1,167) of REV1 protein. Cells were lysed 48 h after transfection and incubated with Ni-NTA resin. Both input lysates and Ni-NTA pulldown were analyzed for FLAG-REV1 and V5 His-REV7 proteins by western blotting with antibodies against FLAG and V5. FLAG-REV1 885–1,167 mutant was highly expressed in cells and a short exposure (sh exp) of this lane in input immunoblot was also shown. (B) Stability of FLAG-REV1 885–1,167 mutant. Cells were transfected with FLAG-REV1 wild-type (wt) or 885–1,167 mutant plasmid. Forty-eight hours after transfection, cells were treated with 15 μg/mL cycloheximide (CHX; lanes 2 and 4) for 12 h before harvest. FLAG-REV1 was analyzed by western blotting. Cyclin B1 was also detected to control for CHX activity. (C) Cells were co-transfected with FLAG-REV1 wild-type (wt) or 885–1167 mutant plus HA-CDH1 plasmids (lanes 2 and 4). Forty-eight hours after transfection, cells were lysed, and FLAG-REV1 and HA-CDH1 proteins were analyzed by western blotting.
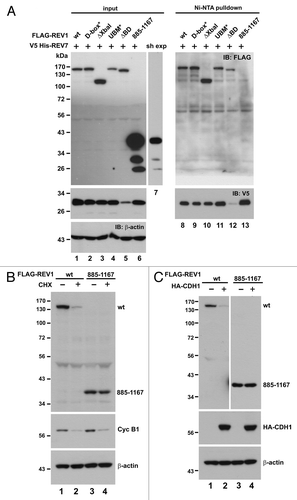
Figure 6. Depletion of REV7 by shRNA stabilizes REV1 protein. (A) Verification of shREV7. HEK293T cells were transfected with increasing amount of plasmid expressing shRNA against REV7 (shREV7, lanes 3–5); a plasmid expressing an irrelevant control shRNA (shCtrl) was used as a negative control. Cells were lysed 48 h after transfection for detection of REV7 and MAD2 protein by western blotting. (B) Cells were co-transfected with FLAG-REV1 plasmid and increasing dose of shREV7 plasmid as indicated for 48 h. Cell lysates were analyzed by western blotting. Expression of FLAG-REV1, cyclin B1 (cyc B1) and REV7 was determined. (C) Knockdown of REV7 prevents REV1 polyubiquitination. Cells were co-transfected with FLAG-REV1 and His-Ub plasmids, plus increasing amount of shREV7 as indicated. Cells transfected with shCtrl was used as a control. Polyubiquitinated REV1 pulled down by Ni-NTA resin was analyzed by western blotting. Expression of REV7 was also determined.
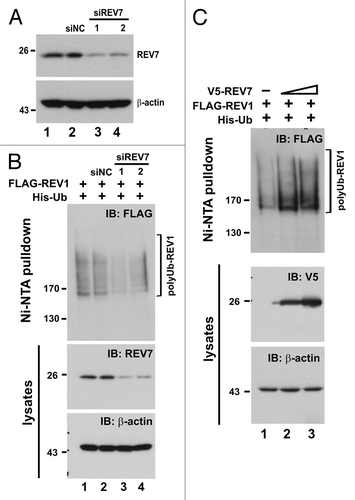
Figure 7. Depletion of REV7 by siRNA stabilizes REV1 protein. (A) Verification of siREV7. Two siRNAs (siREV7-1, -2) targeting two different regions of REV7 were transfected into HEK293T cells for 36 h. siNC, negative control siRNA provided by the manufacturer. (B) Cells were transfected with two siREV7s and siNC separately for 36 h. Then cells were co-transfected with FLAG-REV1 and His-Ub plasmids for 48 h. Ni-NTA resin was added to lysates to pull down polyubiquitinated REV1, which was then analyzed by western blotting. Cells were also lysed with RIPA to detect REV7 protein. (C) Overexpression of REV7 enhances REV1 polyubiquitination. Cells were co-transfected with FLAG-REV1 and His-Ub plasmids, plus increasing amount of V5-REV7 plasmid. Empty V5 vector was also transfected as control (lane 1). Cell lysates were incubated with Ni-NTA resin to pull down polyubiquitinated REV1.