Figures & data
Figure 1. DNA damage response of PA-1 cells to ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and assessed at the indicated time point. Cells were pelleted, cytospun, fixed and stained for (A) pCHK2 (red) or (B) γH2AX (red) and RAD51 (green) in combination with DAPI (blue). Bar = 10 µM. (A) NT PA-1 cells show very faint background pCHK2 staining in non-dividing cells and increased staining of centrosomes in dividing cells (white arrowheads). On day 2 post-ETO, small pCHK2-positive foci accumulate in the nuclei of all cells. By day 4, some small nuclei have lost pCHK2 staining (yellow arrowheads), while large cells remain pCHK2 positive. (B) NT PA-1 cells show no positive staining for γH2AX or RAD51. On day 2, the majority of cells show accumulation of γH2AX or RAD51 positive foci in the nuclei. (C) Cells were cytospun, fixed and stained for DNA image cytometry. DNA content was determined for at least 200 cells in each condition and represented as a percentage. Profound G2 arrest on day 2 was observed followed by the simultaneous emergence of a polyploid (> 4C) and G1 fraction on day 5 before the recovery of the normal cell cycle profile by day 7. (D) The proportion of pCHK2-positive cells was examined in the context of DNA content with cells sub-divided into small or large cells. In the NT control sample, all were pCHK2-negative with an expected nuclei size distribution (2C 80%; ≥ 4C 20%). On day 2, all cells were pCHK2-positive and the vast majority of nuclei were large (≥ 4C). By day 5, cells with small nuclei appear, all of which are pCHK2-negative. Data are representative of > three independent experiments.
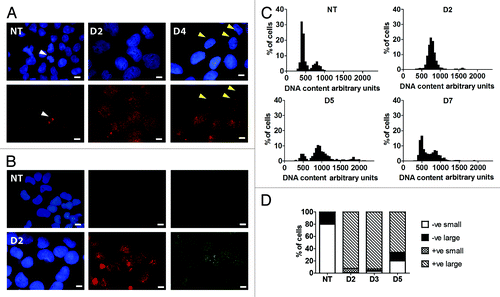
Figure 5. Effect of silencing TP53 on the cell cycle response of PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and examined at the indicated time points by (A and B) flow cytometry, (C) DNA image cytometry and (D) mitotic counts. (A) Both control plasmid and shRNA p53-silenced cells undergo a G2M arrest after ETO treatment, which is, however, more profound from day 3 in TP53-silenced cells. The TP53-silenced cells also show a larger increase in the ≥ 4C cell fraction in response to ETO treatment. Conversely, the recovering G1-2C fraction is reduced. By day 5, the relative proportion of the G2 fraction is diminished in both samples, due to the increase in apoptosis. (B) The proportion of cells in G2 was enumerated over the time course in three independent experiments with the mean and SEM plotted (* p < 0.05). (C) DNA image cytometry analysis was also performed on day 4 after ETO treatment. Apoptotic cells were excluded from the measurements, and highlighted more clearly the relationship between the increased 4C fraction and relative delay in recovery of the proliferating G1-2C fraction (reduced 2.5-fold) after TP53-silencing. The corresponding mitotic index (MI) at this time point is also shown. For full cell cycle dynamics over the time course in this experiment, please see Fig. S6. Data representative of two similar experiments. (D) Mitotic counts were performed on control plasmid or shRNA p53-silenced cells over time after ETO treatment. The results from three independent experiments show a significant increase in the proportion of mitoses after TP53 silencing (* p < 0.05).
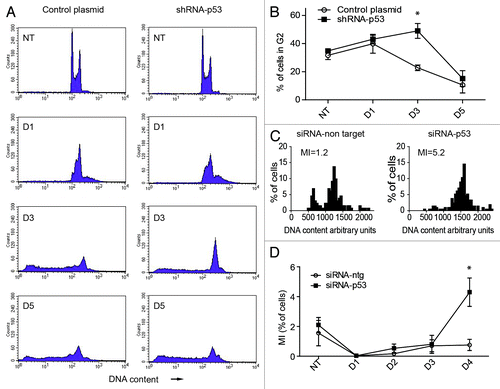
Figure 2. Regulation of TP53, P21CIP1 and OCT4A in PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and assessed at the indicated time point. Cells were pelleted, cytospun, fixed and stained for (A) TP53 (red) (B) P21CIP1 (red) or (C) P21CIP1 (red) and OCT4A (green) in combination with DAPI (blue); Bar = 10 µM. Results representative of nine separate experiments. (A) NT cells showed very faint background staining for TP53. On day 3 post-ETO treatment, TP53 accumulated in the majority of nuclei. (B and C) NT cells had faint background staining for P21CIP1 and weak staining of OCT4A. On day 3 post-ETO treatment, P21CIP1 and OCT4A accumulated in the majority of cell nuclei. (D) Using image analysis on fixed cytospins, the fluorescence intensity of P21CIP1 and OCT4A during the first 3 d post-ETO treatment was determined and plotted for 500 cells in four independent experiments. There was substantial increase and a clear positive correlation between the fluorescent intensity of staining for OCT4A and P21CIP1 after ETO treatment. At the same time, the amplitude of the expression of both factors greatly increased: Non-treated OCT4A expression mean = 2,448, SD = 1,236; OCT4A expression on day 3 mean = 12,641, SD = 6,189; non-treated P21CIP1 expression mean = 2,875, SD = 1,594; P21CIP1 expression on day 3 mean = 10,291, SD = 5,795. (E) Two-channel flow cytometry was used to measure DNA content (PI fluorescence) and OCT4A expression. In NT samples, the OCT4A expression was similar in the 2C and 4C fractions. After ETO treatment, OCT4A was highly elevated in the 4C and 8C fractions and to a lesser extent in the G1-2C fraction. These data indicate that OCT4A accumulation was mostly induced in the DNA damage checkpoint and persisted in the polyploid cells.
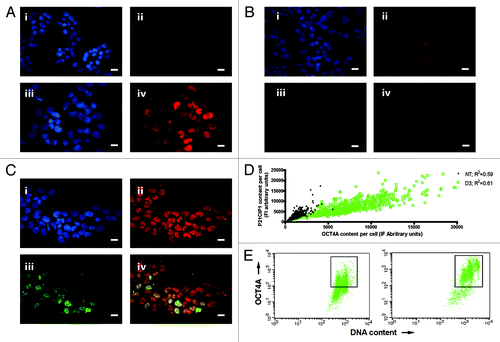
Figure 3. Immunoblot analysis of TP53, OCT4A and P21CIP1 in PA-1 cells after ETO treatment. PA-1 cells were treated with non-target (ntg) siRNA (-) or siRNA-Tp53 (+) for 24 h before treatment with 8 µM ETO, washing after 20 h and cell lysates made and assessed by immunoblotting for Tp53, P21CIP1, OCT4A or GAPDH as a loading control at the indicated time points (day 3 and day 5). Tp53 was upregulated in response to ETO treatment and suppressed by siRNA-TP53. P21CIP1 and OCT4A were also upregulated by ETO treatment, and the upregulation was restricted by treatment with siRNA-TP53. Data are representative of three independent experiments.
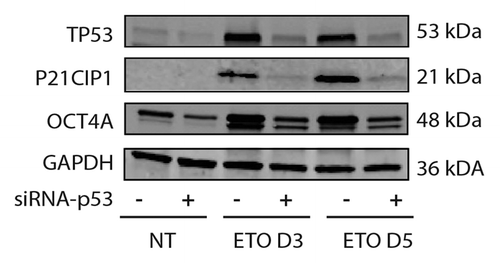
Figure 4. Analysis of OCT4 regulation in PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and examined at the indicated time points by (A) semi-quantitative PCR or (B) cell fractionation and immunoblotting using antibodies against OCT4A or OCT4A and B, actin or histone H3 (H3), the latter two as loading controls. (A) OCT4A transcription was shown to be modestly upregulated in response to ETO after 3 d (upper panel). OCT4B was not substantially detected (middle panel). OCT4B1 was upregulated in response to ETO. OCT4-PG-1 was also upregulated in response to ETO (lower panel). Controls were NTC, non-template control; PBL, peripheral blood lymphocytes. Equivalent amplification of actin after 30 cycles was used as a control for cDNA input (bottom panel). Data representative of three independent experiments. (B) OCT4A protein was upregulated in response to ETO treatment in the cytosol, membrane and nuclear fractions. OCT4A was downregulated in the chromatin-bound fraction. No additional bands were detected with the OCT4A/B dual specificity (C-terminal) antibody compared with the mono-specific OCT4A antibody (N-terminal), indicating only OCT4A species are expressed. Data representative of two independent experiments.
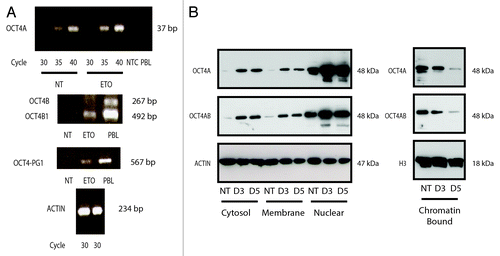
Figure 6. Effect of silencing TP53 on genome instability in PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and examined at the indicated time points for the (A) the proportion of normal (2 centrosomal; 2 cen), ≥ 4 cen bipolar and ≥ 4 cen multipolar mitoses and (B) micronucleation. (A) Cells were treated as before and then examined by IF staining for pCHK2 and α-tubulin. Silencing of TP53 caused multi-centrosomal mitoses (≥ 4 cen) that were mostly multi-polar or showed coalescence of centrosomes in two poles. (B) Cells were treated as before and then assessed for the extent of micronucleation following DNA in situ staining. An increase in the proportion of interphase cells with micronuclei was observed in response to ETO treatment. Silencing of TP53 significantly increased the amount of micronucleation, which was further enhanced in response to ETO (p < 0.05). Data are representative of three independent experiments.
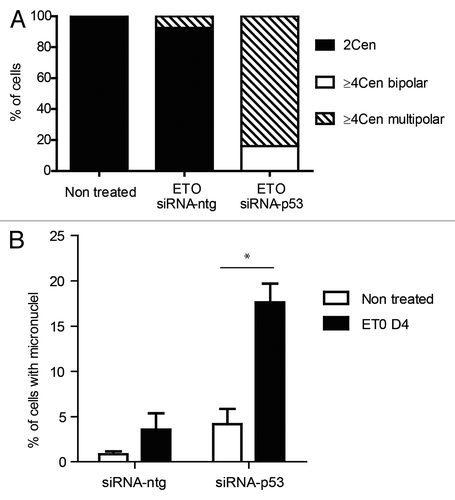
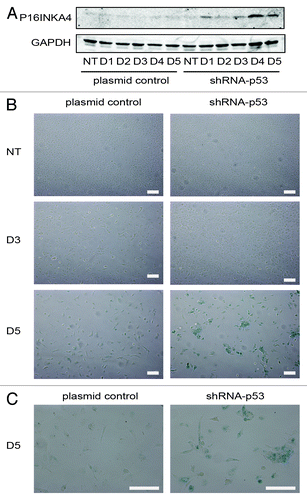
Figure 7. Senescence response of PA-1 cells to ETO treatment. PA-1 cells stably transfected with shRNA-p53 or plasmid control were treated with 8 µM ETO for 20 h, then washed and assessed at the indicated time point by (A) immunoblotting or (B) for sa-β-gal expression and cellular morphology. (A) Cell lysates were made and assessed by immunoblotting for P16INKA4 and GAPDH as a loading control. An accumulation of P16INKA4 was detected in TP53-silenced cells after ETO treatment. (B) sa-β-gal staining was used to detect sa-β-gal activity in response to ETO treatment at indicated time points. Detection of sa-β-gal activity was increased by TP53-silencing and largely mirrored the induction of P16INKA4 seen above. (C) Higher power image of the cells seen on day 5. Bar = 20 µM. Data are representative of > three independent experiments.