Figures & data
Figure 1. (A) STAT5 acetylation-sumoylation-phosphorylation switch. Binding of cytokines to receptors leads to the phosphorylation of STAT5 (P) at tyrosine and serine residues. For example, the interleukins IL-2 and IL-7 induce Janus tyrosine kinases phosphorylating STAT5 at tyrosine residues. The related HATs CBP and p300 catalyze acetylation of STAT5. Phosphorylated STAT5 dimers enter the nucleus and induce STAT5 gene expression promoting cell survival and proliferation. Acetylation (A) of STAT5 rivals sumoylation (S) and thereby allows STAT5 signaling. We postulate that the sumoylation of STAT5 occurs subsequent to a state in which STAT5 is phosphorylated and acetylated, in order to allow gene expression. HDAC9 has been shown to deacetylate STAT5; PTP, phosphatase catalyzing dephosphorylation of STAT5. UBCH9 and PIAS3 belong to the cellular sumoylation machinery (E2/E3 SUMO conjugase/ligase) and transfer SUMO2 to STAT5. SENP1 removes the sumoylation mark and subsequently allows a re-entry of STAT5 into signaling emanating from ligated receptors. The model is based on the works by Ma and colleagues, Van Nguyen and colleagues, and Beier and colleagues (see text for further details). (B) Tyrosine and lysine residues of STAT5 undergoing synergistic and antagonistic posttranslational modifications. The figure shows tyrosine and lysine moieties in STAT5A and STAT5B regulated by phosphorylation, acetylation, and sumoylation. Acetylation and sumoylation of one lysine moiety are mutually exclusive.
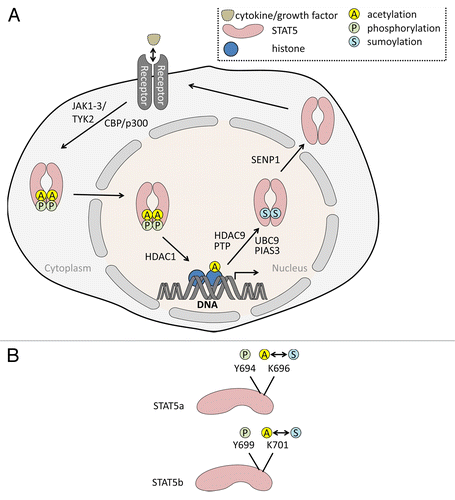
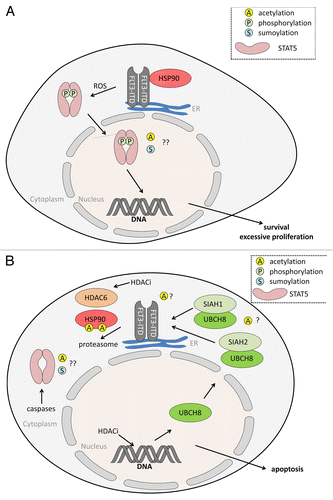
Figure 2. (A) Oncogenic STAT5 activation through FLT3-ITD. FLT3-ITD is a constitutively active, oncogenic driver mutant of FLT3. FLT3-ITD is frequently found in AML and ALL (acute myeloid/lymphoid leukemia). In contrast to FLT3, FLT3-ITD mainly locates to the endoplasmic reticulum (ER), from where it propels STAT5 signaling. Reactive oxygen species (ROS) are involved in the aberrant signaling of FLT3-ITD to STAT5. Resulting gene expression patterns linked to STAT5 promote uncontrolled cell proliferation and apoptosis resistance. It is unknown whether FLT3-ITD-dependent STAT5 activation is linked to the acetylation (A) or sumoylation (S) of STAT5. HSP90 stabilizes FLT3-ITD but is dispensable for the stability of FLT3. For (A) FLT3-ITD is provided as an example for an oncogenic kinase activating STAT5; similar findings were e.g., made for BCR-ABL. (B) HDACi antagonize STAT5 and FLT3-ITD. Histone deacetylase inhibitors (HDACi) eliminate FLT3-ITD and STAT5 via different, partially overlapping molecular mechanisms in AML cells. HDACi induce expression of the UBE2L6 gene encoding the E2 ubiquitin ligase UBCH8. In conjunction with SIAH1 and SIAH2 UBCH8 promotes the poly-ubiquitinylation and proteasomal degradation of FLT3-ITD. Acetylation events may play a role, e.g., HSP90 acetylation occurs in cells treated with HDACi that can block HDAC6. Acetylation of SIAH1, SIAH2, and UBCH8 has so far never been reported. Caspases are activated by HDACi in leukemic cells and these can cleave STAT5. The apoptotic cleavage of STAT5 impairs STAT5-dependent gene expression and this accelerates cellular demise.