Figures & data
Figure 1. (A, B) Schemes illustrating differences between “single epicenter/interaction site model” and “multiple epicenters/interaction sites model.” (A) Single epicenter/interaction interface model: PrPC substrate interacts with PrPSc template at a specific region, irrespective of the strain type, and structural changes spread from this interaction interface to the entire molecule, acting as an epicenter of structural changes. (B) Multiple epicenter/interaction interface model: PrPC substrate can interact with PrPSc template at more than one region and structural changes spread from each epicenter until the entire molecule is converted. (C) Scheme illustrating hypothetic mechanism of dominant-negative inhibition (DNI) when conversion -competent and -incompetent PrPC coexist. The interaction interface of PrPC is represented by a “blue ball.” The blue and red arrows indicate situations where the competition for PrPSc template was won by conversion-competent or conversion-incompetent PrPC, respectively. At the beginning, molecules with an intact interaction interface can bind the PrPSc template irrespective of their conversion abilities, i.e., conversion-competent PrP, “A,” or conversion-incompetent PrP with a defect outside the interface, “B,” can bind, whereas conversion-incompetent PrP with a defect in the interface, “C,” cannot even interact. After binding, “A” converts to a nascent PrPSc “ASc,” while “B” undergoes regional structural changes to become “B*” for high-affinity binding. The PrPSc template bound by “B*” cannot function as the template anymore, consequently inhibiting conversion of “A.”
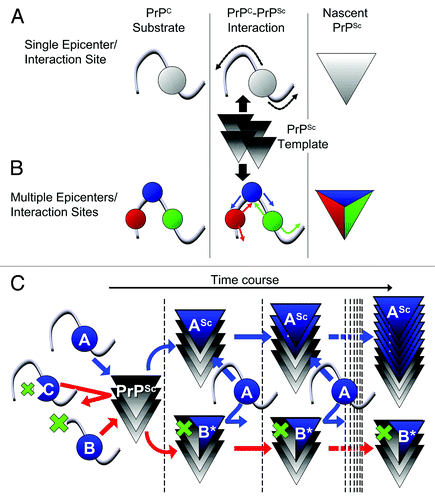
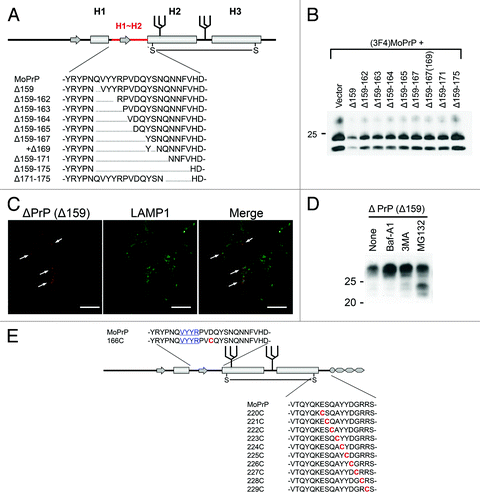
Figure 2. (A) Example of systematically-designed mutant PrPs with deletions in H1~H2, used to test in DNI assay whether the H1~H2 region is an interaction interface. (B) Representative immunoblot demonstrating inverse correlation between DNI efficiency and size of deletion in H1~H2. A conversion-competent and epitope-tagged wild-type PrP, (3F4)MoPrP, and a conversion-incompetent PrP mutant were co-transfected into 22L prion-infected N2a cells and PK-resistant PrP was detected. (C) Confocal image showing co-localization of PrPΔ159 and LAMP1. N2a cells transfected with PrPΔ159 were fixed, treated with 6M guanidine hydrochloride to remove excessive PrPΔ159 signal localized in ER, labeled with antibodies against PrP and LAMP1, and analyzed for co-localization. (D) Immunoblot showing that degradation of PrPΔ159 is inhibited by treatment with bafilomycin A1 (Baf-A1), autophagy inhibitor 3MA, or proteasome inhibitor MG132. (E) A schematic illustration of a series of mutant PrPs with two cysteine substitutions.