Figures & data
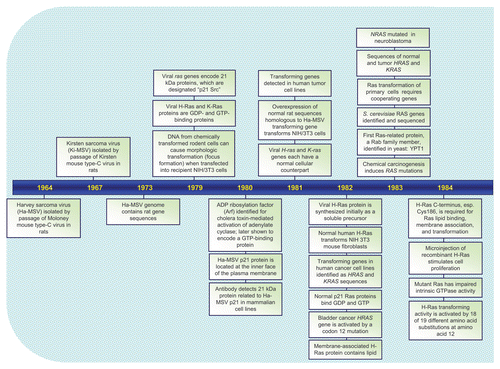
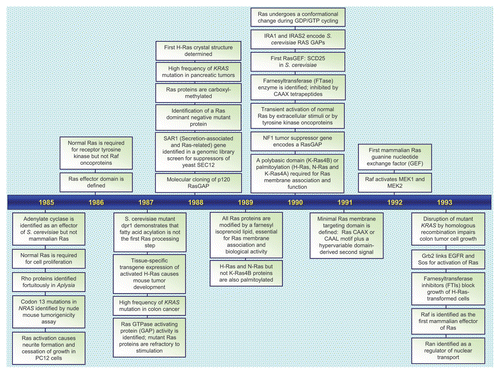
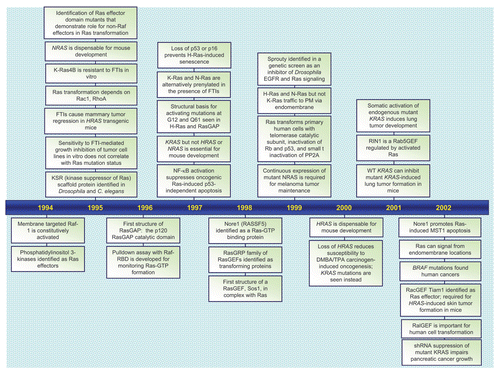
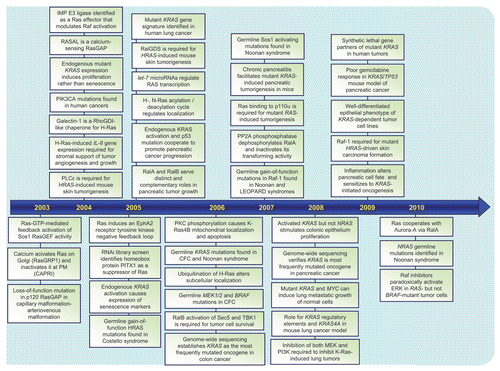
Figure 2 Ras is a GDP/GTP-regulated binary switch. (A) The three RAS genes encode four 188–189 amino acid proteins that share 82–90% overall sequence identity; KRAS encodes two splice variants due to alternative exon 4 utilization, leading to divergent C-terminal sequences. Exons 4A and 4B encode 39 and 38 amino acids, respectively, with 19 identical and 4 conserved substitutions. K-Ras4A is most similar to viral K-ras while K-Ras4B is the predominant isoform expressed in human cells. Residues 1–164 comprise the G domain that contains six conserved sequence motifs shared with other Ras superfamily and GTP-binding proteins. These motifs are involved in binding either phosphate/Mg2+ (PM) or the guanine base (G) of GDP and GTP. Residues in Switch I (aa 30–38) and II (aa 60–76) change in conformation during GDP/GTP cycling. The core effector binding domain (E; residues 32–40) and flanking sequences are involved in effector binding specificity. (B) Regulators of the Ras GDP/GTP cycle. RasGEFs stimulate GDP/GTP exchange. With the 10-fold higher cellular concentrations of GTP over GDP, the net result of RasGEF stimulation is formation of active Ras-GTP. Ras-GTP binds preferentially to downstream effectors. RasGAPs accelerate the intrinsic GTP hydrolysis activity of Ras to promote formation of inactive Ras-GDP. Shown are “classic” missense mutants of Ras proteins that have been useful for dissection of Ras function. The Ras(S17N) dominant negative sequesters and blocks RasGEF activity, preventing Ras activation. The G12V and Q61L mutations, found in human cancers, impair GAP-stimulated GTP hydrolysis. The T35S, E37G and Y40C effector domain mutants (EDMs) differentially impair effector binding. The T35S mutant retains efficient binding to Raf but not PI3K or RalGEF, whereas the E37G mutant retains efficient binding to RalGEF but not Raf or PI3K, and the Y40C mutant retains efficient binding to PI3K but not Raf or RalGEF.
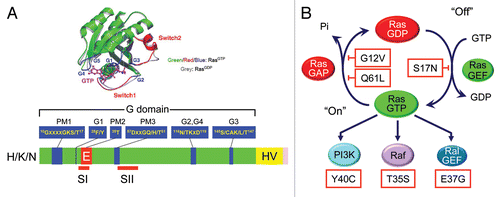
Figure 3 Detection of activated and mutated HRAS in human EJ/T24 bladder carcinoma cells. The NIH/3T3 focus formation assay was used to detect activated oncogenes present in human tumor but not normal genomic DNA. High molecular weight DNA was isolated from the EJ/T24 human bladder carcinoma cell line, converted to a calcium phosphate precipitant, and added to the growth medium of a monolayer of NIH/3T3 cells. After 14 days, foci of morphologically and growth transformed cells can be detected in cultures treated with DNA from tumor cells but not in parallel cultures treated with DNA from normal human cells. The active HRAS fragment from EJ/T24 bladder cells lies within a 4.6 kDa XhoI-SphI fragment. Human H-Ras protein is encoded by sequences spanning four exons. Exon 1 encodes amino acids 1–37. Sequence comparison of the bladder carcinoma-derived HRAS DNA identified a single base substitution at codon 12, resulting in a single missense mutation (G12V).
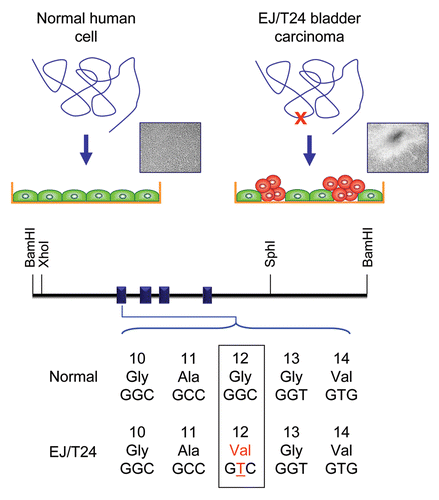
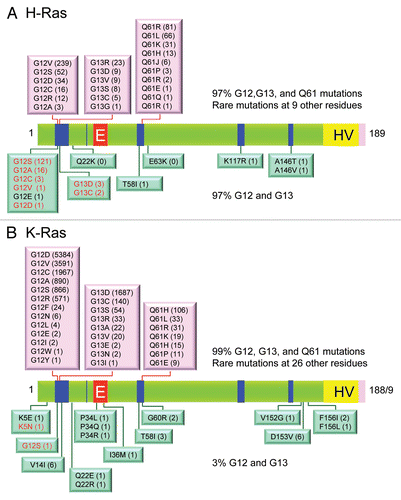
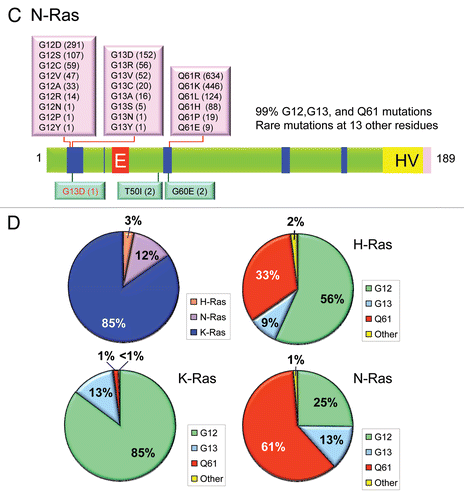
Figure 5 Colorectal and pancreatic cancer progression. (A) Colorectal cancer progression and gene mutations. Colonic epithelial cells undergo a histologic transition from normal to malignant state that is driven by specific genetic events including inactivation of tumor suppressors (APC, SMAD4 and TP53) and activation of the KRAS oncogene.Citation323 The three stages of adenomas represent tumors of increasing size, dysplasia, and villous content. (B) Pancreatic cancer progression and gene mutations. Multiple tumor types arise from the exocrine pancreas, of which greater than 95% are pancreatic ductal adenocarcinoma (PDAC). Normal ductal epithelium progression to infiltrating cancer (left to right) is illustrated through a series of histologically defined precursor lesions (PanINs) that show increasing degrees of disruption of cellular morphology, nuclear atypia and dysplastic growth.Citation324 High grade PanIN-3 progresses to invasive PDAC. Activating point mutations in the KRAS gene occur early, inactivation of the p16/INK4A gene occurs at an intermediate stage, and inactivation of the TP53, SMAD4/DPC4 and BRCA2 genes occurs relatively late. Oncogenes are indicated in green text and tumor suppressors in red text.
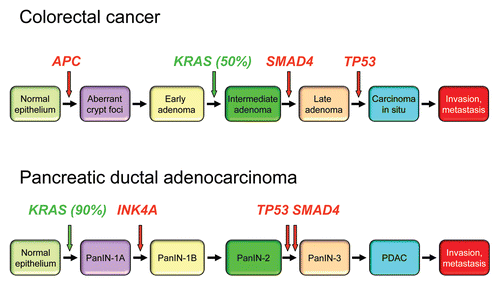
Figure 6 Conservation of Ras proteins in evolution. (A) Domain architecture of Ras proteins and the boundaries of the constant G domains were determined using SMART (http://smart.embl-heidelberg.de/). (B) Clustal/W was then used to align the G domain sequences, and the percent of amino acid identity was determined and used to generate the dendrogram.
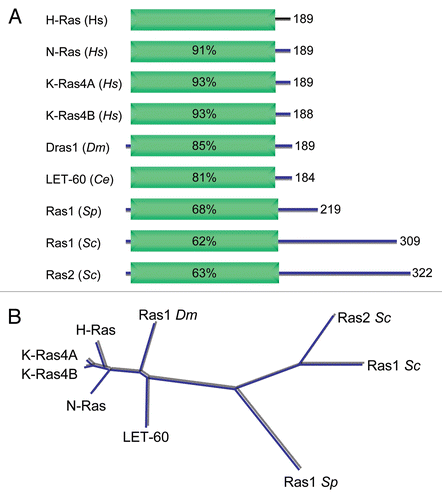
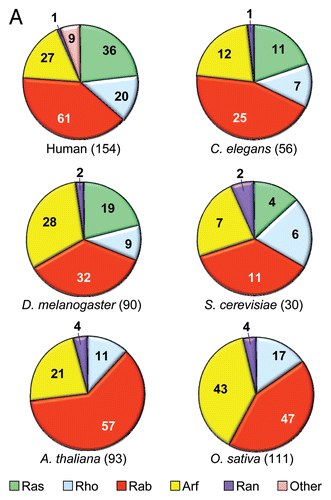
Figure 8 Ras interactome. Proteins that regulate Ras GDP/GTP cycling, catalyze posttranslational modification, or serve as immediate downstream effectors are indicated. Compiled in part from Table 1 in reference Citation121.
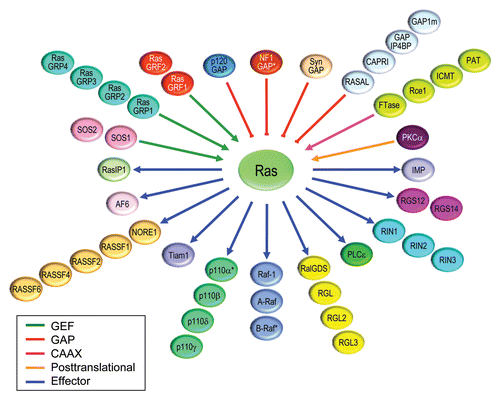

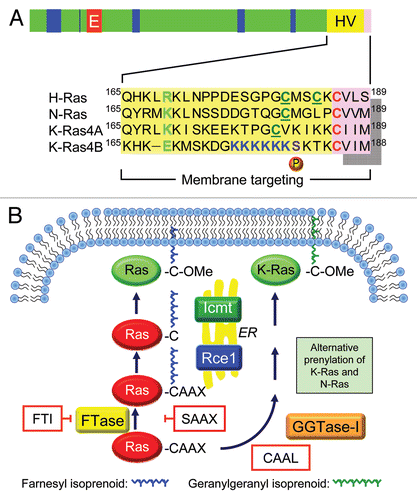
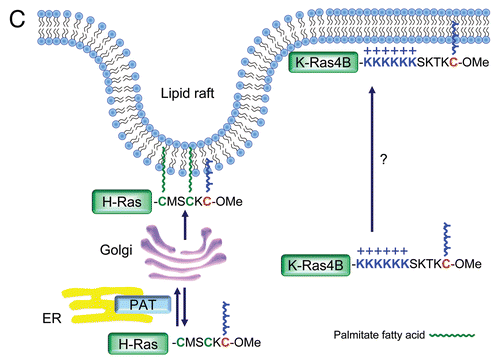
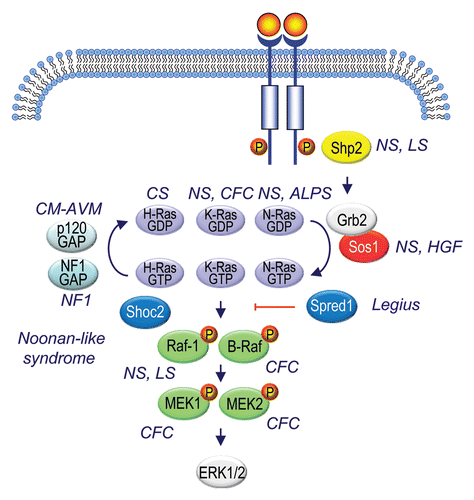
Figure 11 Ras signaling components mutated in RASopathies. Generated based on information summarized in Suppl. Table 3 and from references cited in reference Citation5.