Figures & data
Table 1. Emerging microvascular barrier-enhancing agents
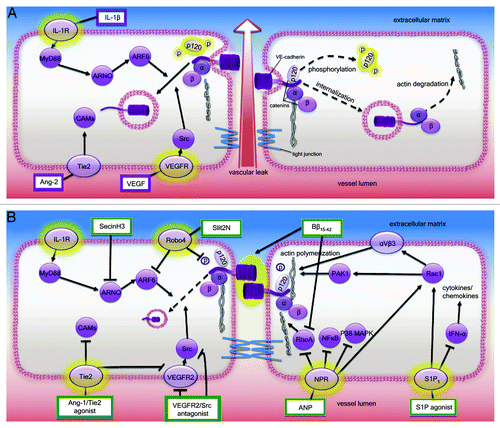
Figure 1. (A) Endothelial cell barrier dysfunction. During severe infections, endothelial activation and dysfunction may lead to the loss of microvascular endothelial barrier integrity, resulting in edema, multiple organ failure and death. Phosphorylation of adherens junction protein p120 catenin precipitates VE-cadherin internalization/junction disassembly. Endothelial integrity may also be compromised by rearrangements/degradation of the actin cytoskeleton. Molecular pathways implicated in this response include MYD88-ARNO binding that results in enhanced ARF6 signaling and decreased VE-cadherin localization at the cell surface. Endothelial activation may also cause an increase in the Ang-1 antagonist, Ang-2. Ang-2 binds to its cognate receptor, Tie2, and impedes the vascular stabilizing effects of Ang-1 by promoting proinflammatory endothelial responses and upregulating cell surface adhesion molecules. Vascular permeabilizing VEGF binds to VEGFR2 resulting in increased dissociation of VE-cadherin from the adherens junction through a VEGFR2-Src-VE-cadherin signaling pathway. (B) Endothelial cell barrier enhancement. SecinH3, a GEF (e.g., ARNO) inhibitor, inhibits ARF6-induced VE-cadherin internalization. Ang-1/Tie2 agonists activate Tie2 resulting in increased vascular quiescence via strengthening of endothelial cell junctions, downregulation of surface adhesion molecules and transdominate blockade of VEGR2 signaling. Fibrinopeptide Bβ15–42 provides barrier protection via maintenance of membrane VE-cadherin and inhibition of actin degradation via RhoA signaling inhibition. Upon binding its receptor Robo4, Slit2N reduces p120 catenin phosphorylation and inhibits ARF6, thereby increasing VE-cadherin retention at the cell surface. ANP administration decreases microvascular permeability via inhibition of RhoA-induced actin degradation and NFκB/P38 MAPK inhibition. S1P agonist administration reinforces the endothelial barrier via Rac1 and αvβ3 integrin signaling, resulting in formation and stabilization of cortical actin. Administration of S1P agonist enhances cortical actin formation (via Rac1 and αvβ3 integrin signaling) and downregulates IFN-α, thereby decreasing cytokine/chemokine production and enhancing endothelial cell barrier stability.