
Abstract
Mycobacterium avium subsp. paratuberculosis (MAP) is the causative agent of Johne’s disease (JD) which affects mainly ruminants and is characterized by chronic diarrhea and emaciation. Johne’s disease is highly prevalent in many countries around the world and leads to high economic losses associated with decreased production. Genotyping of the involved pathogen could be used in the study of population genetics, pathogenesis and molecular epidemiology including disease surveillance and outbreak investigation. Principally, researchers have first assumed the presence of two different MAP strains that are associated with the animal host species (cattle and sheep). However, nowadays MAP characterization depends mainly upon genetic testing using genetic markers such as insertion elements, repetitive sequences and single nucleotide polymorphisms. This work aims to provide an overview of the advances in molecular biological tools used for MAP typing in the last two decades, discuss how these methods have been used to address interesting epidemiological questions, and explore the future prospects of MAP molecular epidemiology given the ever decreasing costs of the high throughput sequencing technology.
Introduction
Mycobacterium (M.) avium subsp. paratuberculosis (MAP) is a member of the M. avium complex (MAC). It is the causative agent of paratuberculosis or Johne’s disease (JD), a chronic gastroenteritis primarily affecting domestic ruminants and causing high economic losses especially in the dairy industry worldwide [Citation1]. The diasease was also found to sporadically exist in wild ruminants [Citation2]. MAP was also identified – but without showing clinical signs – in a wide range of hosts such as non-human primates [Citation3], non-ruminant wildlife [Citation4], dogs [Citation5], feral cats [Citation6], rabbits [Citation7], parrots [Citation8] and bears [Citation9]. Over a century, a possible role for MAP in the pathogenesis of Crohn’s diseases (CD), a chronic debilitating gastroenteritis affecting humans has been debated. One of the first MAP isolates that was obtained from a Crohn’s disease patient is the “Linda strain” [Citation10]. The difficulty of constant and reliable MAP isolation from human CD patients was one of the main obstacles that hindered the proof or refutation of this hypothesis over such a long period of time [Citation11,Citation12].
Molecular epidemiology of infectious diseases is a branch of epidemiology that involves the use of molecular biological techniques in studying the determinants and distribution of disease occurrence in a certain population [Citation13]. In this review, we will focus on the current understanding of MAP diversity, the advances achieved in the molecular biological approaches of MAP genotyping in the last two decades and the epidemiological applications of these approaches.
MAP types
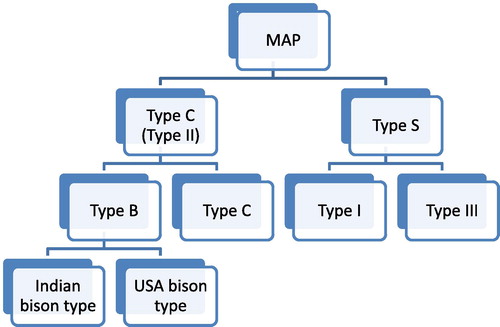
Historically, MAP types were described based on epidemiologic and phenotypic characteristics. Based on the host from which MAP strains were first isolated, researchers proposed cattle (C) and sheep (S) type strains as the two main MAP types infecting cattle and sheep, respectively [Citation14]. However, advances in the molecular biology have provided a more robust basis for classification. Using pulsed-field gel electrophoresis (PFGE), two MAP genetic groups were identified that showed only host preferences but not exclusivity and hence a new nomenclature for MAP types (type I and II) has been proposed [Citation15]. Type I isolates were slow growers (more than 16 weeks to achieve visible growth) and strongly associated with sheep; while type II isolates were readily growing (4–16 weeks) strains commonly isolated from cattle, but with a broader host range. A third group called “intermediate” or “type III” was also characterized and was primarily thought to be different from type S and type C isolates [Citation16]. However, a recent whole genome sequencing-based study revealed that both types (I and III) are subgroups of the sheep or type S [Citation17].
Based on a SNP at bp 223 of the insertion element IS1311, a new MAP type termed “bison” or “type B” was described for isolates originating from bisons (Bison bison) in Montana, USA [Citation18]. All IS1311 copies in “type B” isolates had a thymidine transition compared to cytosine in type S isolates. In type C isolates, some copies possessed T and others C nucleotides (nt). Whole genome sequencing analysis later showed that the type B is a subgroup of type C and that some type C strains exhibited the IS1311 profile of type S raising concerns about the reliability of IS1311 analysis as a method of choice to discriminate between types C, S, and B [Citation17]. Subsequent molecular analysis of Indian MAP type B isolates revealed variations from those isolated in the USA and consequently respective isolates were termed “Indian bison type” [Citation19]. Later, the Indian bison type isolates were found to possess a TG deletion at bp positions 64 and 65 of the IS1322 locus 2 [Citation20]. The current classification of MAP types based on a recent whole genome sequencing-based study [Citation17] is illustrated in .
Genotyping methods
Many genotyping methods have been developed and applied for MAP characterization. In , the different methods described in details in this review are listed.
able 1 MAP genotyping methods described in this review.
.1 Randomly amplified polymorphic DNA (RAPD)
RAPD analysis is a simple and cost effective PCR-based fingerprinting technique. In this analysis, the genomic DNA is directly amplified under low stringency PCR-conditions using a single short primer (10–22 bases) of arbitrary sequence. In the last decade of the 20th century, RAPD was commonly used for subtyping of different organisms such as Mycoplasma [Citation28], M. tuberculosis [Citation29] and M. avium [Citation30]. In Germany, RAPD was used for the first time to characterize MAP isolates (n = 16) from cattle [Citation21]. The isolates displayed heterogeneity; however the data was insufficient to conclude that RAPD could be applied in MAP genotyping. More recently, one primer -among 20 different primers available in a commercial kit- was found to be suitable for the identification and subtyping of MAP and M. avium using RAPD analysis [Citation31]. Six different genotypes were identified for both MAP and M. avium isolates investigated. MAP isolates from cattle, goat and humans were genetically similar, while one sheep isolate had a distinct genetic profile.
.2 Amplified fragment length polymorphism (AFLP)
AFLP is a rapid PCR-based fingerprinting technique. It involves DNA fragmentation with the help of restriction enzymes followed by ligation of adaptors that are complimentary to the restriction regions. The adapted restriction fragments are then amplified with PCR and visualized on polyacrylamide gels either by autoradiography or fluorescence based methods. A variety of restriction enzymes and adaptors/primers could be combined making AFLP a flexible tool that for different applications such as genetic characterization and genetic mapping. Moreover, AFLP is sensitive to polymorphism detection at the whole genome level.
In one study in the USA, AFLP was used to characterize 104 genetically diverse MAP isolates obtained from different hosts and geographic regions [Citation22]. In contrast to earlier studies, AFLP fingerprints of the MAP isolates obtained from humans did not cluster with either the bovine or ovine isolates [Citation22,Citation32]. Among the studied MAP isolates, bovine isolates showed a low degree of genetic diversity regardless of geographic origin, while isolates obtained from humans and sheep displayed a higher degree of genetic heterogeneity. Contrarily, another American research group found a high degree of genetic diversity among MAP isolates characterized by AFLP using 96 primer set (eleven genotypes for 21 isolates; [Citation33]). More recently, the same research group reported that AFLP could even discriminate between MAP isolates based on epigenetic variations [Citation34]. Despite no sequence differences were detected, MAP isolates obtained from tissue samples possessed distinct AFLP fingerprints compared with isolates cultured from faecal samples. Interestingly, they were able to identify restriction sites that were not digested in the tissue-associated isolates and thus accounting for the apparent heterogeneity. The authors assumed that this could be due to the presence of a DNA sequence they identified upstream of the undigested sites for possible methyltransferase recognition.
AFLP suffers however from some limitations such as the need for a purified, high molecular weight DNA, poor reproducibility especially between different platforms and difficulty of standardization due to a subjective interpretation of the banding patterns [Citation35].
.3 Pulsed-field gel electrophoresis (PFGE)
PFGE is a fingerprinting-based genotyping technique that was first described by Schwartz et al. in 1984 for the separation of yeast chromosomes [Citation36]. However, it is still the gold standard for characterizations of many pathogenic bacteria [Citation37]. PFGE is a special form of gel electrophoresis where restricted DNA fragments are subjected to periodical reorientation of the electric field relative to the gel direction allowing a better separation of DNA fragments [Citation38]. In Australia, PFGE was used for the first time for genotyping of MAP isolates obtained from different ruminant species [Citation23]. PFGE was able to discriminate between the three main MAP types (I, II and III). As a first step, it has clustered pigmented and non-pigmented MAP isolates into two genetic groups (type I and type II) [Citation15]. Type III MAP isolates were then later found to have distinct PFGE fingerprinting profiles [Citation16].
PFGE was not commonly used in the literature as a MAP typing technique. Few studies have used PFGE for characterization of MAP isolates originating from different European countries [Citation39–Citation42].
.4 IS900-restriction fragment length polymorphisms (IS900-RFLP)
RFLP is a fingerprinting-based typing approach that involves digestion of an organism’s genome using one or more restriction endonuclease enzymes followed by electrophoresis of DNA fragments. These fragments are then transferred to a membrane (“Southern blot”), where they are hybridized with a labelled DNA probe targeting the MAP-specific insertion sequence (IS900). IS900-RFLP utilizes the random nature of IS900 insertion sites that differ from one MAP strain to another. Consequently, upon hybridization each MAP strain would exhibit different banding patterns [Citation43]. Most studies have used one or more of the three enzymes BstEII, PvuII and PstI. However, combining the results of two or more IS900-RFLP reactions employing different restriction enzymes increases the overall discriminatory power/index (DI) of MAP genotyping [Citation44]. DI is a numerical value ranging from 0 to 1 which measures the probability that a certain typing method would discriminate between two epidemiologically unrelated strains of a particular microbial population [Citation45]. IS900-RFLP was also able to differentiate between the three major MAP types “type I or S”, “type II or C” and “type III or intermediate” [Citation14].
IS900-RFLP was the first and the most commonly used MAP genotyping approach for almost two decades [Citation41,Citation44,Citation46–Citation48]. Nevertheless, it has drawbacks limiting its use especially after the advancement of sequencing technologies in the last years. It is laborious, time consuming, requires a substantial amount of cultured material for genomic DNA extraction [Citation49] and the inter-laboratory interpretation of results might be challenging.
.5 Tandem repeats-based genotyping
Tandem repeats (micro- or minisatellites) are DNA sequences that are repeated in a head to tail manner. They are common in prokaryotic genomes and the repeat copy number may vary even between closely related strains [Citation50]. Based on the repeat size, tandem repeats are classified into three main categories. Repeats ranging from 1 to 9 nt are called microsatellites, short sequence repeats (SSR), or short tandem repeats. Larger repeats (10–100 nt) are usually known as minisatellites or variable number tandem repeats (VNTR), while repeats of more than 100 nt are termed macrosatellites [Citation51]. Two mechanisms were hypothesized as the drivers for the addition or deletion of repeat copies, namely strand slippage caused by DNA polymerase errors on one side and recombination events on the other side [Citation50]. Tandem repeats were first thought to be junk DNA of no function; however, an accumulating body of evidence has shown that they affect the expression of genes associated with bacterial adaptation to the surrounding environment [Citation52]. Epidemiologists have exploited the mutable nature of such repeats to characterize bacterial pathogens based on the polymorphism that results from variations in repeat copy numbers [Citation53]. The characterization is founded on PCR-based amplification of tandem repeat loci followed by calculating the repeat copy numbers at each locus either by size polymorphism mainly in gel or capillary electrophoresis (for VNTR) or by sequencing the PCR products (for SSR) [Citation54,Citation55]. The repeat copy numbers at different tandem repeat loci are then combined together in a numerical code defining the genotype of each isolate. As a PCR-based approach, tandem repeats exhibit some advantages compared with the other typing methods. They have made the characterization faster and less laborious. Furthermore, they enabled direct typing from clinical specimens or from non-viable cultures. Other advantages are reproducibility, standardization, and comparability of results between laboratories [Citation25].
.5.1 Mycobacterial interspersed repetitive unit-variable number tandem repeat
Mycobacterial interspersed repetitive units (MIRU) are a specific group of minisatellites found only in mycobacteria. They were first described in the genome of M. tuberculosis [Citation56]. Compared with other minisatellites (VNTR), MIRU have a unique structure of two DNA sequence motifs; A (24 bp) 5′-TGACGAGGAGCGGCGCAGATGGCA-3′ and B (29 bp) 5′-GGCGCCGGTGACGATGCAGAGCGTAGCGA-3′ [Citation57]. Since both types of repeats (MIRU and VNTR) have already been used for MAP characterization in the majority of studies, the nomenclature MIRU-VNTR was established in the field.
MIRU loci (MIRU 1-MIRU 4) were first described in MAC strains by nucleotide basic local alignment search tool (BLASTN) analysis of the previously identified MIRU sequences in M. tuberculosis [Citation24]. These loci were able to discriminate between MAP and other MAC subspecies and between pigmented ovine and all other MAP isolates as well. One year later [Citation58], VNTR loci were investigated in the publicly available complete genome of M. avium strain 104 (GenBank accession number http://ncbi-n:NC_008595.1) using the tandem repeat finder software [Citation59]. By investigating 49 MAP isolates, eight VNTR candidates were tested and five of them were found to be polymorphic dividing the isolates into six genotypes. One of these five VNTR loci (VNTR 1658) was indeed identical to the previously described MIRU 3 locus [Citation24]. In 2007, a set of eight MIRU-VNTR loci (VNTR 3, 7, 10, 25, 32, 47 and MIRU X3 and 292) was discovered in the MAP genome [Citation60]. MIRU X3 and 292 are the same loci as the previously described MIRU 3 and 2, respectively [Citation24]. These eight loci have been used to set up an online database (available at http://mac-inmv.tours.inra.fr/index.php) to facilitate the documentation and comparison of MIRU-VNTR-based MAP genotypes detected in different laboratories all over the world. This has encouraged many researchers to use this MIRU-VNTR panel to carry out epidemiological investigations for MAP isolates from different countries [Citation42,Citation44,Citation47,Citation61–Citation66]. In 2010, a new VNTR locus (VNTR 259) was described which was able to discriminate between MAP type II and type III isolates [Citation67].
MIRU-VNTR loci of MAP were recently found to be a subject of homoplasy and hence compromising their accuracy as genetic markers for phylogenetic analysis [Citation68]. Homoplasy is the independent development of the same alleles in isolates belonging to different genetic lineages of an organism based on convergent evolution [Citation17].
.5.2 Multi-locus short sequence repeats (MLSSR)
In a first screening of SSR in the MAP genome, an American research group identified 78 perfect mono-, di- and tri-nucleotide repeats by in silico analysis of the first complete genome of MAP strain K10 [Citation25]. Perfect repeats are characterized by an integer number of identical copies of the original repeat sequence, whereas imperfect repeats may possess a sequence of n integer copies plus a fragment of the original repeat sequence. In a preliminary analysis using a subset of diverse MAP isolates (n = 6), eleven of these repeats were found to be polymorphic (SSR 1-SSR 11) [Citation25]. In a sequencing-based approach, these polymorphic loci were subsequently utilized for the typing of 27 genetically diverse MAP isolates. The analysis exhibited a high DI of 0.96 by identifying 20 different SSR types (two major phylogenetic groups) which was sufficient to support epidemiological investigation. Shortly afterwards, American research groups started to use this genotyping approach to characterize MAP isolates originating from different host species and geographical regions in the USA [Citation69–Citation73].
Furthermore, the stability of SSR loci as genetic markers was investigated. In one study, SSR markers remained unchanged (stable) after ten in vitro serial passages of three genetically distinct MAP isolates [Citation73]. Contrarily, SSR locus 2 exhibited a repeat copy number variation both after in vivo passage and in a natural infection context within single cattle herds and individual animals as well [Citation74], hence it was recommended to refrain from using this locus in epidemiological investigations due to its instability. Based on technical limitations of sequencing a long stretch of a mononucleotide repeat, it was proposed that more than ten guanine bases at SSR locus 1 should be interpreted as ≥11 G [Citation74], which limits the discriminatory capacity of this locus. Different attempts were carried out to overcome this problem by determination of the copy number at this locus either by Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF MS; [Citation75]) or by capillary gel electrophoresis-based fragment analysis [Citation76]. This in turn extended the detection limit of copy numbers at SSR locus 1 to 15 and 21 nt, respectively.
.6 Combination of different genotyping markers
Bacteria are described to be monomorphic, when they exhibit very low genomic diversity. Some important pathogens such as M. tuberculosis, Bacillus anthracis, Yersinia pestis and MAP are monomorphic [Citation77]. With respect to MAP genotyping, different authors therefore used combinations of two or more genotyping techniques to increase the overall typing DI in order to obtain epidemiologically meaningful data ().
able 2 Different combinations of genotyping markers previously used for MAP typing.
.7 Single nucleotide polymorphisms (SNP)
Single nucleotide polymorphisms (SNPs) are point mutations of single nt along the genome. They are stable genetic markers that can be used to trace back the phylogenetic history of an organism. The availability of the first complete MAP genome [Citation27] has directed the scientists’ efforts to search for SNPs that could be used for MAP typing and phylogenetic analyses.
Investigation of single or multiple gene sequences such as 65 kDa heat shock protein (hsp65), superoxide dismutase (sodA), chromosomal replication initiator protein (dnaA), DNA polymerase III subunit beta (dnaN), DNA replication and repair protein (recF), DNA topoisomerase IV subunit B (gyrB), enoyl-(acyl carrier protein) reductase (inhA), polyketide synthase (pks8), DNA gyrase subunit A (gyrA) and PPE family protein (MAP1506) enabled scientists to find SNPs differentiating between the major MAP types (I, II and III) [Citation26,Citation85–Citation87]. Partial sequencing of IS900, a robust and merely MAP-specific insertion element, revealed SNP characteristics for the MAP types I, II, and III adding a typing feature to the well-established diagnostic value of this element [Citation88]. The emergence of new technologies allowed researchers to adopt novel approaches such as microarray technology and high resolution melting (HRM) for detection of the previously described MAP-specific SNPs [Citation67,Citation89]. Recently, the decrease in the costs of high throughput sequencing (HTS) has made the whole genome sequencing (WGS) of bacteria more accessible to microbiologists. Accordingly, MAP isolates have been sequenced and informative SNPs were used for developing assays that can be used to address various epidemiological questions. For example, seven SNPs were derived from WGS data of human MAP isolates and used to investigate the occurrence of identical lineages of human MAP isolates in other hosts in Australia [Citation90,Citation91]. Similarly, five SNPs were extracted from WGS data of a national Canadian collection of cattle MAP isolates [Citation92,Citation93] and were used to detect the four most common phylogenetic groups in Canada. The preliminary climax was reached, when a comprehensive SNP-based MAP typing assay was developed based on an international collection of MAP isolates for the first time [Citation94]. This was feasible by extracting only 14 SNPs that have been used to classify MAP isolates into 14 different phylogenetic groups.
.8 Whole genome sequencing (WGS)
Using a shotgun sequencing approach, the first closed and annotated genome sequence of the MAP cattle strain K10 was published in 2005 [Citation27], which has later been revisited and improved [Citation95]. This has facilitated subsequent MAP genetic studies for diagnostic, comparative genomic and epidemiological purposes. It also gave insight into the molecular basis of MAP pathogenicity and virulence characteristics. Since then, researchers have worked on reinforcing the scientific knowledge by publishing complete genomes of other MAP isolates from different geographic areas. Currently, 44 genomes are publicly available in the genome database (https://www.ncbi.nlm.nih.gov/genome/genomes) of the National Center for Biotechnology Information (NCBI) for MAP isolates obtained from different countries such as India, Egypt, Germany, and the USA [Citation96–Citation98].
The recent application of HTS enabled researchers to investigate the MAP sequence variations at the whole genome level. Consequently, comparative genomic studies were carried out to gain insight into genomic variations between different MAP types obtained from several host species including sheep [Citation99], camel [Citation100], bison [Citation101], human [Citation90,Citation102] and other domestic and wild animals [Citation103]. The exponential decrease in the costs of HTS has enabled the WGS of a large number of MAP isolates. This helped elucidating phylogeography of MAP isolates at a national level in Canada [Citation68,Citation92] and – in the meantime – also on a worldwide scale [Citation17].
Epidemiological applications of MAP genotyping
In this section, examples of the application of MAP genotyping in addressing interesting epidemiological aspects of JD will be discussed.
.1 Within-herd and within-animal strain diversity
Using a wide spectrum of typing techniques, multiple MAP strains were identified in the same herd and even in the same animal [Citation32,Citation39,Citation40,Citation47,Citation62,Citation71–Citation74,Citation76,Citation104–Citation108]. Different explanations were proposed in the literature for the observed multiple strain superinfections. Since animals’ movements are well known as an important risk factor for inter-herd MAP transmission [Citation109], multiple introduction events associated with purchasing subclinically infected animals from different sources is the most popular and likely the most plausible explanation [Citation44,Citation72]. However, one study has assumed that polyclonality could be a basic aspect of the organism’s ecology and has proposed that clinical cases of paratuberculosis might be provoked by multiple MAP-strain infections [Citation62]. A third explanation was that MAP strains exhibiting minor genetic diversity could exist due to within-herd or within-animal evolution over time (microevolution) given the chronic nature of the organism [Citation107]. The extent of microevolution in MAP infections and its possible phenotypic consequences need to be further investigated. Moreover, the potential consequences of multiple strain infections on the clinical outcome, immune reaction, and vaccination efficacy should be addressed in upcoming studies.
.2 Informing paratuberculosis management policies
One of the most important epidemiological applications of genotyping is tracing back sources of infections and hence providing invaluable information for decision makers concerning disease management policies [Citation110]. In this regard, a high resolution genotyping that could classify strains in the greatest number of groups is necessary for an efficient tracing back process [Citation111].
MAP isolates obtained from cattle showing clinical signs of JD in Queensland, a subtropical Australian area known to have a very low prevalence of JD, were subjected to genotyping using a combination of MIRU-VNTR and MLSSR [Citation112]. The genotypes were compared with national and international strains to identify the potential source(s) of infection and to evaluate the applied control measures. The results indicated that the two identified MAP strains (Q2012 and Q2013) resulted from two different incursion events and were not introduced from other Australian regions, where the disease is more prevalent. Although no definitive clarification of the sources of infections was achieved, the findings of this study provided evidence that restriction of animal movements between high- and low-prevalence regions as a control measure applied has not been violated in Australia.
.3 Revealing the inter-species MAP transmission
MAP genotypic characterization has helped elucidating MAP transmission events between different host species. Cross-species transmission between cattle and different wildlife animal species has been reported. Using a combination of three genotyping approaches (SSR, MIRU-VNTR, and IS900-RFLP), a German study investigated the transmission of MAP between free-living red deer and farmed cattle sharing the same habitat [Citation47]. Four of the identified 17 genotypes were shared between the two animal species, where one genotype was found to dominate in both of them suggesting a cross-species transmission between cattle and deer [Citation47]. In Australia, a small proportion (1.7%) of macropods co-grazing with sheep was found to be infected with MAP [Citation113]. Genotypic characterization of the isolates using PCR and restriction endonuclease analysis has indicated that a cross-species transmission of the MAP type S has occurred. However, macropods seems not to be a wildlife reservoir of infection, since they were found to shed the organism at low levels [Citation113]. On the other side, wild rabbits are believed to represent a reservoir of MAP infection [Citation114] and a genotyping approach combining IS900-RFLP, PFGE, and MIRU-VNTR has revealed that wild rabbits and cattle within the same region often share the same genotypes [Citation42]. Moreover, MAP isolates obtained from CD patients showed a great homogeneity (based on WGS) with bovine MAP isolates from the same geographic location in Australia indicating a possible zoonotic transmission, presumably through consumption of milk and dairy products [Citation90].
Conclusions
The advances in the molecular biological techniques have revolutionized our understanding of the MAP diversity and the epidemiology of JD has witnessed a shift from depending on classical phenotypic traits such as host preference, time to visible growth and colony pigmentation for MAP characterization towards using genotyping approaches. The latter have so far successfully been used for interesting epidemiological applications. This included informing paratuberculosis management policies by identifying the sources of infections, detection of multiple MAP strains both at the intra-herd and intra-animal levels and revealing the inter-species MAP transmission. In the near future, HTS technology is expected to be more accessible to more laboratories throughout the world. Consequently, WGS will transform our ability to characterize MAP isolates at an unprecedented high resolution which would be applied to extend our understanding of MAP molecular epidemiology at intra-animal, intra-herd, regional, national and international levels.
Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Competing interests
The authors have no competing interests to declare.
Notes
Peer review under responsibility of Faculty of Veterinary Medicine, Cairo University.
References
- W.C.LosingerEconomic impact of reduced milk production associated with Johne's disease on dairy operations in the USAJ Dairy Res7242005425432
- M.MachackovaP.SvastovaJ.LamkaI.ParmovaV.LiskaJ.SmolikParatuberculosis in farmed and free-living wild ruminants in the Czech Republic (1999–2001)Vet Microbiol10142004225234
- K.FechnerK.Matz-RensingK.LampeF.J.KaupC.P.CzernyJ.SchaferDetection of Mycobacterium avium subsp. paratuberculosis in non-human primatesJ Med Primatol2017
- M.FlorouL.LeontidesP.KostoulasC.BillinisM.SofiaI.KyriazakisIsolation of Mycobacterium avium subspecies paratuberculosis from non-ruminant wildlife living in the sheds and on the pastures of Greek sheep and goatsEpidemiol Infect13652008644652
- M.A.MillerS.C.DaveyL.S.VAn HeldenF.KettnerS.M.WeltanR.LastParatuberculosis in a domestic dog in South AfricaJ S Afr Vet Assoc882017
- M.V.PalmerW.C.StoffregenJ.G.CarpenterJ.R.StabelIsolation of Mycobacterium avium subsp. paratuberculosis (MAP) from feral cats on a dairy farm with MAP-infected cattleJ Wildl Dis4132005629635
- E.A.RaizmanS.J.WellsP.A.JordanG.D.DelGiudiceR.R.BeyMycobacterium avium subsp. paratuberculosis from free-ranging deer and rabbits surrounding Minnesota dairy herdsCan J Vet Res69120053238
- P.MünsterK.FechnerI.VölkelA.von BuchholzC.P.CzernyDistribution of Mycobacterium avium ssp. paratuberculosis in a German zoological garden determined by IS900 semi-nested and quantitative real-time pcrVet Microbiol1631–22013116123
- M.KopecnaS.OndrusI.LiterakJ.KlimesA.HorvathovaM.MoravkovaDetection of Mycobacterium avium subsp. paratuberculosis in two brown bears in the central European CarpathiansJ Wildl Dis4232006691695
- R.J.ChiodiniH.J.Van KruiningenW.R.ThayerR.S.MerkalJ.A.CoutuPossible role of mycobacteria in inflammatory bowel disease. I. An unclassified Mycobacterium species isolated from patients with Crohn's diseaseDig Dis Sci2912198410731079
- L.A.SechiC.T.DowMycobacterium avium ss. Paratuberculosis zoonosis – the hundred year war – beyond Crohn's diseaseFront Immunol6201596
- A.FawzyA.PrinceA.A.HassanA.FayedM.ZschöckM.NagaEpidemiological studies on Johne’s disease in ruminants and Crohn’s disease in humans in EgyptInt J Vet Sci Med1220137986
- B.FoxmanL.RileyMolecular epidemiology: Focus on infectionAm J Epidemiol15312200111351141
- D.M.CollinsD.M.GabricG.W.De LisleIdentification of two groups of Mycobacterium paratuberculosis strains by restriction endonuclease analysis and DNA hybridizationJ Clin Microbiol282199015911596
- K.StevensonV.M.HughesLd.JuanN.F.InglisF.WrightJ.M.SharpMolecular characterization of pigmented and nonpigmented isolates of Mycobacterium avium subsp. paratuberculosisJ Clin Microbiol405200217981804
- Ld.JuanA.MateosL.DomínguezJ.M.SharpK.StevensonGenetic diversity of Mycobacterium avium subspecies paratuberculosis isolates from goats detected by pulsed-field gel electrophoresisVet Microbiol1063–42005249257
- J.M.BryantV.C.ThibaultD.G.SmithJ.McLuckieI.HeronI.A.SevillaPhylogenomic exploration of the relationships between strains of Mycobacterium avium subspecies paratuberculosisBMC Genomics17201679
- R.J.WhittingtonI.B.MarshR.H.WhitlockTyping of is 1311 polymorphisms confirms that bison (Bison bison) with paratuberculosis in Montana are infected with a strain of Mycobacterium avium subsp. paratuberculosis distinct from that occurring in cattle and other domesticated livestockMol Cell Probes1532001139145
- I.SevillaS.V.SinghJ.M.GarridoG.AdurizS.RodriguezM.V.GeijoMolecular typing of Mycobacterium avium subspecies paratuberculosis strains from different hosts and regionsRev Sci Technol243200510611066
- J.S.SohalS.V.SinghP.K.SinghA.V.SinghN.KumarA new marker is1311 l2 PCR-REA for identification of 'Indian bison' type Mycobacterium avium subspecies paratuberculosisIndian J Biotechnol122013204207
- P.ScheiblG.F.GerlachDifferentiation of Mycobacterium paratuberculosis isolates by rDNA-spacer analysis and random amplified polymorphic DNA patternsVet Microbiol572–31997151158
- A.S.MotiwalaM.StrotherA.AmonsinB.ByrumS.A.NaserJ.R.StabelMolecular epidemiology of Mycobacterium avium subsp. paratuberculosis: evidence for limited strain diversity, strain sharing, and identification of unique targets for diagnosisJ Clin Microbiol415200320152026
- M.M.FeizabadiI.D.RobertsonA.HopeD.V.CousinsD.J.HampsonDifferentiation of Australian isolates of Mycobacterium paratuberculosis using pulsed-field gel electrophoresisAust Vet J75121997887889
- T.J.BullK.Sidi-BoumedineE.J.McMinnK.StevensonR.PickupJ.Hermon-TaylorMycobacterial interspersed repetitive units (MIRU) differentiate Mycobacterium avium subspecies paratuberculosis from other species of the Mycobacterium avium complexMol Cell Probes1742003157164
- A.AmonsinL.L.LiQ.ZhangJ.P.BannantineA.S.MotiwalaS.SreevatsanMultilocus short sequence repeat sequencing approach for differentiating among Mycobacterium avium subsp. paratuberculosis strainsJ Clin Microbiol424200416941702
- C.Y.TurenneM.SemretD.V.CousinsD.M.CollinsM.A.BehrSequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complexJ Clin Microbiol4422006433440
- L.LiJ.P.BannantineQ.ZhangA.AmonsinB.J.MayD.AltThe complete genome sequence of Mycobacterium avium subspecies paratuberculosisProc Natl Acad Sci USA1023520051234412349
- B.R.CharltonA.A.BickfordR.P.ChinR.L.WalkerRandomly amplified polymorphic DNA (RAPD) analysis of Mycoplasma gallisepticum isolates from turkeys from the central valley of CaliforniaJ Vet Diagn Invest1151999408415
- J.P.SinghR.VermaP.ChaudhuriRandom amplified polymorphic DNA (RAPD) analysis of Mycobacterium tuberculosis strains in IndiaJ Vet Sci722006181187
- M.SchrenzelM.NicolasC.WitteR.PapendickT.TuckerL.KeenerMolecular epidemiology of Mycobacterium avium subsp. avium and Mycobacterium intracellulare in captive birdsVet Microbiol1261–32008122131
- S.R.PillaiB.M.JayaraoJ.D.GummoE.C.HueD.TiwariJ.R.StabelIdentification and sub-typing of Mycobacterium avium subsp. paratuberculosis and Mycobacterium avium subsp. avium by randomly amplified polymorphic DNAVet Microbiol7932001275284
- R.J.WhittingtonA.F.HopeD.J.MarshallC.A.TaragelI.MarshMolecular epidemiology of Mycobacterium avium subsp. paratuberculosis: IS900 restriction fragment length polymorphism and IS1311 polymorphism analyses of isolates from animals and a human in AustraliaJ Clin Microbiol389200032403248
- B.O'SheaS.KhareK.BlissP.KleinT.A.FichtL.G.AdamsAmplified fragment length polymorphism reveals genomic variability among Mycobacterium avium subsp. paratuberculosis isolatesJ Clin Microbiol428200436003606
- B.O'SheaS.KhareP.KleinA.RousselL.G.AdamsT.A.FichtAmplified fragment length polymorphism reveals specific epigenetic distinctions between Mycobacterium avium subspecies paratuberculosis isolates of various isolation typesJ Clin Microbiol496201122222229
- S.BenschM.AkessonTen years of aflp in ecology and evolution: why so few animals?Mol Ecol1410200528992914
- D.C.SchwartzC.R.CantorSeparation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresisCell37119846775
- D.MacCannellBacterial strain typingClin Lab Med3332013629650
- R.V.GoeringPulsed field gel electrophoresis: a review of application and interpretation in the molecular epidemiology of infectious diseaseInfect Genet Evol1072010866875
- I.SevillaJ.M.GarridoM.GeijoR.A.JustePulsed-field gel electrophoresis profile homogeneity of Mycobacterium avium subsp. paratuberculosis isolates from cattle and heterogeneity of those from sheep and goatsBMC Microbiol7200718
- I.SevillaL.LiA.AmonsinJ.M.GarridoM.V.GeijoV.KapurComparative analysis of Mycobacterium avium subsp. paratuberculosis isolates from cattle, sheep and goats by short sequence repeat and pulsed-field gel electrophoresis typingBMC Microbiol82008204
- Z.Dimareli-MalliK.StevensonK.SarrisK.SossidouStudy of microbiological and molecular typing aspects of paratuberculosis in sheep and goats in Northern GreeceTransbound Emerg Dis566–72009285290
- K.StevensonJ.AlvarezD.BakkerF.BietL.D.JuanS.DenhamOccurrence of Mycobacterium avium subspecies paratuberculosis across host species and European countries with evidence for transmission between wildlife and domestic ruminantsBMC Microbiol92009212
- I.PavlikA.HorvathovaL.DvorskaJ.BartlP.SvastovaR.du MaineStandardisation of restriction fragment length polymorphism analysis for Mycobacterium avium subspecies paratuberculosisJ Microbiol Methods381999155167
- P.MöbiusG.LuyvenH.HotzelH.KöhlerHigh genetic diversity among Mycobacterium avium subsp. paratuberculosis strains from german cattle herds shown by combination of IS900 restriction fragment length polymorphism analysis and mycobacterial interspersed repetitive unit-variable-number tandem-repeat typingJ Clin Microbiol4632008972981
- H.GrundmannS.HoriG.TannerDetermining confidence intervals when measuring genetic diversity and the discriminatory abilities of typing methods for microorganismsJ Clin Microbiol3911200141904192
- L.Abdolmohammadi KhiavM.HaghkhahK.TadayonN.MosavariGenotyping analysis of bovine, ovine, and caprine paratuberculosis in Iran: an IS900-RFLP studyInt J Mycobacteriol5Suppl. 12016S228
- I.FritschG.LuyvenH.KöhlerW.LutzP.MöbiusSuspicion of Mycobacterium avium subsp. paratuberculosis transmission between cattle and wild-living red deer (Cervus elaphus) by multitarget genotypingAppl Environ Microbiol784201211321139
- V.F.SaundersG.J.EamensM.J.TurnerT.M.JessepIdentification of a new RFLP type of Mycobacterium avium subsp. paratuberculosis in epidemiological tracing of bovine Johne’s diseaseAust Vet J812003564566
- A.S.MotiwalaL.LiV.KapurS.SreevatsanCurrent understanding of the genetic diversity of Mycobacterium avium subsp. paratuberculosisMicrobes Infect85200614061418
- Z.SunW.LiS.XuH.HuangThe discovery, function and development of the variable number tandem repeats in different Mycobacterium speciesCrit Rev Microbiol4252016738758
- K.ZhouA.AertsenC.W.MichielsThe role of variable DNA tandem repeats in bacterial adaptationFEMS Microbiol Rev3812014119141
- A.JansenR.GemayelK.J.VersterpenUnstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequencesGenome Dyn72012108125
- C.S.ChiouMultilocus variable-number tandem repeat analysis as a molecular tool for subtyping and phylogenetic analysis of bacterial pathogensExpert Rev Mol Diagn10201057
- A.El-SayedS.NaturN.-E.M.I.AbdouM.SalemA.HassanM.ZschöckGenotyping of Mycobacterium avium field isolates based on repetitive elementsInt J Vet Sci Med1120133642
- M.SalemS.NaturA.A.El-SayedA.HassanG.BaljerM.ZschöckMolecular characterization of Mycobacterium avium subsp. paratuberculosis field isolates recovered from dairy cattle in GermanyInt J Vet Sci Med1120133035
- P.SupplyJ.MagdalenaS.HimpensC.LochtIdentification of novel intergenic repetitive units in a mycobacterial two-component system operonMol Microbiol2619979911003
- E.CastellanosL.D.JuanL.DomínguezA.AranazProgress in molecular typing of Mycobacterium avium subspecies paratuberculosisRes Vet Sci9222012169179
- P.OverduinL.SchoulsP.RohollA.van der ZandenN.MahmmodA.HerreweghUse of multilocus variable-number tandem-repeat analysis for typing Mycobacterium avium subsp. paratuberculosisJ Clin Microbiol4211200450225028
- G.BensonTandem repeats finder: a program to analyze DNA sequencesNucl Acids Res271999573580
- V.C.ThibaultM.GrayonM.L.BoschiroliC.HubbansP.OverduinK.StevensonNew variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS900 and IS1245 restriction fragment length polymorphism typingJ Clin Microbiol458200724042410
- F.BietI.A.SevillaT.CochardL.H.LefrançoisJ.M.GarridoI.HeronInter- and intra-subtype genotypic differences that differentiate Mycobacterium avium subspecies paratuberculosis strainsBMC Microbiol122012264
- H.GerritsmannG.L.StalderJ.SpergserF.HoelzlA.DeutzA.Kuebber-HeissMultiple strain infections and high genotypic diversity among Mycobacterium avium subsp. paratuberculosis field isolates from diseased wild and domestic ruminant species in the Eastern Alpine region of AustriaInfect Genet Evol212014244251
- A.GioffréM.Correa MuñozM.F.Alvarado PinedoR.VacaC.MorsellaM.A.FiorentinoMolecular typing of Argentinian Mycobacterium avium subsp. paratuberculosis isolates by multiple-locus variable number-tandem repeat analysisBraz J Microbiol4622015557564
- Z.RónaiÁ.CsivincsikM.GyuraneczZ.KreizingerÁ.DánS.JánosiMolecular analysis and MIRU-VNTR typing of Mycobacterium avium subsp. paratuberculosis strains from various sourcesJ Appl Microbiol11822015275283
- B.R.ImperialeR.D.MoyanoA.B.Di GiulioM.A.RomeroM.F.Alvarado PinedoM.P.SantangeloGenetic diversity of Mycobacterium avium complex strains isolated in Argentina by MIRU-VNTREpidemiol Infect1457201713821391
- Md.KruijfO.N.LesniakD.YearsleyE.RamovicA.CoffeyJ.O'MahonyLow genetic diversity of bovine Mycobacterium avium subspecies paratuberculosis isolates detected by MIRU-VNTR genotypingVet Microbiol2032017280285
- E.CastellanosB.RomeroS.RodríguezLd.JuanJ.BezosA.MateosMolecular characterization of Mycobacterium avium subspecies paratuberculosis types II and III isolates by a combination of MIRU-VNTR lociVet Microbiol1441–22010118126
- C.AhlstromH.W.BarkemaK.StevensonR.N.ZadoksR.BiekR.KaoLimitations of variable number of tandem repeat typing identified through whole genome sequencing of Mycobacterium avium subsp. paratuberculosis on a national and herd levelBMC Genomics162015161
- A.H.GhadialiM.StrotherS.A.NaserE.J.ManningS.SreevatsanMycobacterium avium subsp. paratuberculosis strains isolated from Crohn's disease patients and animal species exhibit similar polymorphic locus patternsJ Clin Microbiol4211200453455348
- A.S.MotiwalaA.AmonsinM.StrotherE.J.B.ManningV.KapurS.SreevatsanMolecular epidemiology of Mycobacterium avium subsp. paratuberculosis isolates recovered from wild animal speciesJ Clin Microbiol424200417031712
- A.S.MotiwalaM.StrotherN.E.TheusR.W.StichB.ByrumW.P.ShulawRapid detection and typing of strains of Mycobacterium avium subsp. paratuberculosis from broth culturesJ Clin Microbiol435200521112117
- A.K.PradhanR.M.MitchellA.J.KramerM.J.ZurakowskiT.L.FyockR.H.WhitlockMolecular epidemiology of Mycobacterium avium subsp. paratuberculosis in a longitudinal study of three dairy herdsJ Clin Microbiol4932011893901
- N.B.HarrisJ.B.PayeurV.KapurS.SreevatsanShort-sequence-repeat analysis of Mycobacterium avium subsp. paratuberculosis and Mycobacterium avium subsp. avium isolates collected from animals throughout the United States reveals both stability of loci and extensive diversityJ Clin Microbiol448200629702973
- N.KasnitzH.KöhlerM.WeigoldtG.F.GerlachP.MöbiusStability of genotyping target sequences of Mycobacterium avium subsp. paratuberculosis upon cultivation on different media, in vitro- and in vivo passage, and natural infectionVet Microbiol1673–42013573583
- C.AhlstromH.W.BarkemaJ.D.BuckImproved short-sequence-repeat genotyping of Mycobacterium avium subsp. paratuberculosis by using matrix-assisted laser desorption ionization-time of flight mass spectrometryAppl Environ Microbiol8022014534539
- M.P.PodderS.E.BanfieldG.P.KeefeH.G.WhitneyK.TahlanTyping of Mycobacterium avium subspecies paratuberculosis isolates from Newfoundland using fragment analysisPLoS One1042015 e0126071
- I.ComasS.HomolkaS.NiemannS.GagneuxGenotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologiesPLoS One4112009 e7815
- A.El-SayedA.A.HassanS.NatourA.AbdulmawjoodM.BülteW.WolterEvaluation of three molecular methods of repetitive element loci for differentiation of Mycobacterium avium subsp. paratuberculosis (MAP)J Microbiol4732009253259
- P.E.DouarreW.CashmanJ.BuckleyA.CoffeyJ.O'MahonyMolecular characterization of Mycobacterium avium subsp. paratuberculosis using multi-locus short sequence repeat (MLSSR) and mycobacterial interspersed repetitive units-variable number tandem repeat (MIRU-VNTR) typing methodsVet Microbiol1493–42011482487
- J.A.Fernández-SilvaA.AbdulmawjoodÖ.AkinedenM.BülteGenotypes of Mycobacterium avium subsp. paratuberculosis from South American countries determined by two methods based on genomic repetitive sequencesTrop Anim Health Prod446201211231126
- J.A.Fernández-SilvaA.AbdulmawjoodÖ.AkinedenK.DrägerW.KlawonnM.BülteMolecular epidemiology of Mycobacterium avium subsp. paratuberculosis at a regional scale in GermanyRes Vet Sci9322012776782
- J.B.OkuniC.I.DovasP.LoukopoulosI.G.BouzalasD.P.KateeteM.L.JolobaIsolation of Mycobacterium avium subspecies paratuberculosis from Ugandan cattle and strain differentiation using optimised DNA typing techniquesBMC Vet Res8201299
- J.S.SohalJ.ArsenaultO.LabrecqueJ.-H.FairbrotherJ.-P.RoyG.FecteauGenetic structure of Mycobacterium avium subsp. paratuberculosis population in cattle herds in quebec as revealed by using a combination of multilocus genomic analysesJ Clin Microbiol528201427642775
- P.R.HunterM.A.GastonNumerical index of the discriminatory ability of typing systems: an application of simpson's index of diversityJ Clin Microbiol2611198824652466
- E.CastellanosA.AranazB.RomeroLd.JuanJ.AlvarezJ.BezosPolymorphisms in gyra and gyrb genes among Mycobacterium avium subsp. paratuberculosis type I, II, and III isolatesJ Clin Microbiol4510200734393442
- I.B.MarshR.J.WhittingtonGenomic diversity in Mycobacterium avium: single nucleotide polymorphisms between the S and C strains of M. avium subsp. paratuberculosis and with M. a. aviumMol Cell Probes21120076675
- T.A.GriffithsK.RiouxJ.De BuckSequence polymorphisms in a surface PPE protein distinguish types I, II, and III of Mycobacterium avium subsp. paratuberculosisJ Clin Microbiol464200812071212
- E.CastellanosA.AranazL.de JuanJ.AlvarezS.RodriguezB.RomeroSingle nucleotide polymorphisms in the IS900 sequence of Mycobacterium avium subsp. paratuberculosis are strain type specificJ Clin Microbiol477200922602264
- M.GastaldelliE.StefaniA.A.LettiniPozzato N. Multiplexed typing of Mycobacterium avium subsp. paratuberculosis types I, II, and III by luminex xmap suspension arrayJ Clin Microbiol4912011389391
- J.W.WynneT.J.BullT.SeemannD.M.BulachJ.WagnerC.D.KirkwoodExploring the zoonotic potential of Mycobacterium avium subspecies paratuberculosis through comparative genomicsPLoS One672011 e22171
- J.W.WynneC.BellerV.BoydB.FrancisJ.GwozdzM.CarajiasSNP genotyping of animal and human derived isolates of Mycobacterium avium subsp. paratuberculosisVet Microbiol1723–42014479485
- C.AhlstromH.W.BarkemaK.StevensonR.N.ZadoksR.BiekR.KaoGenome-wide diversity and phylogeography of Mycobacterium avium subsp. paratuberculosis in Canadian dairy cattlePLoS One1122016e0149017
- C.AhlstromH.W.BarkemaJ.De BuckRelative frequency of 4 major strain types of Mycobacterium avium ssp. paratuberculosis in Canadian dairy herds using a novel single nucleotide polymorphism-based polymerase chain reactionJ Dairy Sci9910201682978303
- C.LeaoR.J.GoldstoneJ.BryantJ.McLuckieJ.InacioD.G.E.Smithsingle nucleotide polymorphism-based assay for genotyping Mycobacterium avium subsp. paratuberculosisJ Clin Microbiol5432016556564
- J.W.WynneT.SeemannD.M.BulachS.A.CouttsA.M.TalaatW.P.MichalskiResequencing the Mycobacterium avium subsp. paratuberculosis k10 genome: Improved annotation and revised genome sequenceJ Bacteriol19223201063196320
- A.S.AminC.Y.HsuS.F.DarwishP.GhoshE.M.AbdEl-FatahT.S.BehourEcology and genomic features of infection with Mycobacterium avium subspecies paratuberculosis in EgyptMicrobiol161Pt 42015807818
- J.P.BannantineL.LiM.MwangiR.CoteJ.A.Raygoza GarayV.KapurComplete genome sequence of Mycobacterium avium subsp. paratuberculosis, isolated from human breast milkGenome Announc2014;2(1).
- P.MöbiusM.HölzerM.FelderG.NordsiekM.GrothH.KöhlerComprehensive insights in the Mycobacterium avium subsp. paratuberculosis genome using new WGS data of sheep strain JIII-386 from GermanyGenome Biol Evol2015evv154
- J.P.BannantineC.-W.WuC.HsuS.ZhouD.C.SchwartzD.O.BaylesGenome sequencing of ovine isolates of Mycobacterium avium subspecies paratuberculosis offers insights into host associationBMC Genomics13201289
- P.GhoshC.HsuE.J.AlyamaniM.M.ShehataM.A.Al-DubaibA.Al-NaeemGenome-wide analysis of the emerging infection with Mycobacterium avium subspecies paratuberculosis in the Arabian camels (Camelus dromedarius)PLoS One722012 e31947
- J.S.SohalN.SheoranK.NarayanasamyV.BrahmachariS.SinghS.SubodhGenomic analysis of local isolate of Mycobacterium avium subspecies paratuberculosisVet Microbiol1343–42009375382
- V.J.TimmsK.A.HassanH.M.MitchellB.A.NeilanComparative genomics between human and animal associated subspecies of the Mycobacterium avium complex: a basis for pathogenicityBMC Genomics162015695
- C.Y.HsuC.W.WuA.M.TalaatGenome-wide sequence variation among Mycobacterium avium subspecies paratuberculosis isolates: a better understanding of Johne's disease transmission dynamicsFront Microbiol22011236
- I.PavlikL.BejckovaM.PavlasZ.RozsypalovaS.KoskovaCharacterization by restriction endonuclease analysis and DNA hybridization using IS900 of bovine, ovine, caprine and human dependent strains of Mycobacterium paratuberculosis isolated in various localitiesVet Microbiol451995311318
- D.V.CousinsS.N.WilliamsA.HopeG.J.EamensDNA fingerprinting of Australian isolates of Mycobacterium avium subsp. paratuberculosis using using IS900 RFLPAust Vet J7832000184190
- P.MobiusG.LuyvenH.HotzelH.KohlerHigh genetic diversity among Mycobacterium avium subsp. paratuberculosis strains from German cattle herds shown by combination of IS900 restriction fragment length polymorphism analysis and mycobacterial interspersed repetitive unit-variable-number tandem-repeat typingJ Clin Microbiol4632008972981
- K.J.E.van HulzenH.C.M.HeuvenM.NielenJ.HoeboerW.J.SantemaA.P.KoetsDifferent Mycobacterium avium subsp. paratuberculosis MIRU-VNTR patterns coexist within cattle herdsVet Microbiol1482–42011419424
- F.W.DavidsonC.AhlstromJ.De BuckH.G.WhitneyK.TahlanExamination of Mycobacterium avium subspecies paratuberculosis mixed genotype infections in dairy animals using a whole genome sequencing approachPeerJ42016 e2793
- N.MarquetouxC.HeuerP.WilsonA.RidlerM.StevensonMerging DNA typing and network analysis to assess the transmission of paratuberculosis between farmsPrev Vet Med1342016113121
- A.van BelkumP.T.TassiosL.DijkshoornS.HaeggmanB.CooksonN.K.FryGuidelines for the validation and application of typing methods for use in bacterial epidemiologyClin Microbiol Infect13Suppl. 32007146
- J.P.BannantineL.-L.LiS.SreevatsanV.KapurHow does a mycobacterium change its spots? Applying molecular tools to track diverse strains of Mycobacterium avium subspecies paratuberculosisLett Appl Microbiol5732013165173
- J.OakeyL.GaveyS.V.SinghJ.PlatellD.WaltisbuhlVariable-number tandem repeats genotyping used to aid and inform management strategies for a bovine Johne's disease incursion in tropical and subtropical AustraliaJ Vet Diagn Invest2652014651657
- P.C.ClelandD.R.LehmannP.H.PhillipsD.V.CousinsL.A.ReddacliffR.J.WhittingtonA survey to detect the presence of Mycobacterium avium subspecies paratuberculosis in Kangaroo island macropodsVet Microbiol1453–42010339346
- L.J.ShaughnessyL.A.SmithJ.EvansD.AndersonG.CaldowG.MarionHigh prevalence of paratuberculosis in rabbits is associated with difficulties in controlling the disease in cattleVet J19812013267270