
Abstract
Approximately 10.8% of human cancers are associated with infection by an oncogenic virus. These viruses include human papillomavirus (HPV), Epstein–Barr virus (EBV), Merkel cell polyomavirus (MCV), human T-cell leukemia virus 1 (HTLV-1), Kaposi's sarcoma-associated herpesvirus (KSHV), hepatitis C virus (HCV) and hepatitis B virus (HBV). These oncogenic viruses, with the exception of HCV, require the host RNA splicing machinery in order to exercise their oncogenic activities, a strategy that allows the viruses to efficiently export and stabilize viral RNA and to produce spliced RNA isoforms from a bicistronic or polycistronic RNA transcript for efficient protein translation. Infection with a tumor virus affects the expression of host genes, including host RNA splicing factors, which play a key role in regulating viral RNA splicing of oncogene transcripts. A current prospective focus is to explore how alternative RNA splicing and the expression of viral oncogenes take place in a cell- or tissue-specific manner in virus-induced human carcinogenesis.
Introduction
Infection with human oncogenic viruses is the cause of ∼10.8% of human cancers worldwide.Citation1 The first oncogenic virus to be identified, avian Rous sarcoma virus, was discovered by Peyton Rous in 1911.Citation2 Decades later, a series of other oncogenic viruses were also discovered, including cottontail rabbit papillomavirus,Citation3 mouse mammary tumor virus,Citation4 adenovirus,Citation5,Citation6 and simian virus 40 (SV40).Citation7 The theory of virus-mediated oncogenesis was finally experimentally demonstrated in 1976 by Harold Varmus and Michael Bishop through the identification of v-src as the Rous sarcoma virus viral oncogene.Citation8 Characterization of viral oncogenes also brought numerous landmarks in understanding of the oncogenic process, such as the discoveries of p53 as an SV40 T-antigen-associated proteinCitation9,Citation10 and E2F as a mediator of adenovirus E1A function.Citation11
Although the oncogenic viruses found in the early studies exhibited oncogenic activity in animal cells, they failed to transform human cells. The theory of viral oncogenesis in humans remained controversial until 1965, when Epstein–Barr virus (EBV) was discovered in Burkitt lymphoma cells ().Citation12 Subsequent demonstrations, including isolation of human T-cell lymphoma virus-1 (HTLV-1) from adult T-cell lymphoma (ATL)Citation13,Citation14,Citation15 and the association of high-risk human papillomaviruses (HPVs) with cervical cancers,Citation16,Citation17,Citation18,Citation19 paved the way for the concept of human tumor viruses. Discovery of the association of Kaposi's sarcoma-associated herpes virus (KSHV) with Kaposi's sarcomaCitation20 and lymphoma,Citation21 and of Merkel cell polyomavirus (MCV) with Merkel cell carcinoma (MCC)Citation22 underscores the possibility that even more tumor viruses will be discovered by modern technology. These tumor-inducing human viruses encode viral oncogenes or genes with oncogenic activities and utilize cellular machinery for their expression and the transformation of host cells. One of the indispensable steps for viral oncogene expression is RNA splicing, which is essential for almost all tumor viruses to diversify their transcriptomes during virus infection and oncogenesis. This review will highlight the recent advances in understanding of human viral oncogenes and the importance of RNA splicing in their expression.
Table 1 Oncogenic human viruses and viral oncogenes.
HUMAN PAPILLOMAVIRUS
HPV and oncogenic activity
HPVs are a group of non-enveloped double-stranded DNA viruses that preferentially infect the cells in the basal layer of skin and mucosal tissues, primarily through microtraumas or close contact. More than 180 HPV genotypes have been reported (http://pave.niaid.nih.gov). HPVs associated with cancers are referred to as high-risk HPVs, and those associated with benign anogenital or skin warts are low-risk HPVs.Citation23,Citation24 To date, more than 95% of cervical cancers, 50%–90% of other anogenital cancers and 20%–30% of oropharyngeal cancers have been associated with persistent infection and genomic integration of high-risk HPVs.Citation24,Citation25,Citation26 Among the common high-risk HPVs (HPV16, 18, 31, 33, 45 and 58), HPV16 is the most prevalent genotype and is responsible for ∼60% of cervical cancer cases worldwide.Citation18,Citation26 The HPV16 viral genome encodes eight open reading frames (ORFs). Six (E1, E2, E4, E5, E6 and E7) are encoded from the early region and two (L1 and L2) are encoded from the late region of the virus genome. E6 and E7 are responsible for the oncogenic activities of high-risk HPVs.
The ∼18 kDa HPV16 E6 oncoprotein consists of ∼150 amino acid (aa) residues with four CxxC zinc-binding motifs (Figure ), which form two hypothetical zinc fingers. The C-terminus of HPV16 E6 has a PSD-95/disks large/zonula occludins (PDZ)-binding domain and interacts with several PDZ proteins, such as SAP97/hDlgCitation30,Citation31 and MAGI-1.Citation32,Citation33 HPV16 E6 is targeted to the nucleus through its three nuclear localization signals,Citation34 and its oncogenic activity depends mainly on E6-mediated p53 degradation. E6 from high-risk HPVs, but not from low-risk HPVs, interacts with E3 ubiquitin ligase E6-associated protein (E6AP) and promotes the target recognition of E6AP, which leads to ubiquitination of p53.Citation35 HPV16 E6 appears to be more potent than HPV18 E6 for p53 degradation.Citation35 E6AP binds to E6 through its acidic LxxLL motif and stabilizes the E6 protein.Citation36,Citation37,Citation38 The N-terminal region of HPV16 E6 is responsible for E6 homodimerization, which is necessary for the p53-targeting activity of HPV16 E6.Citation39 The F47 residue within the N-terminal hydrophobic interface of HPV16 E6 is essential, and mutation of this residue results in loss of the p53 degradation activity of E6.Citation39,Citation40,Citation41 E6 also participates in multiple oncogenic events through protein–protein interactions. E6 interacts with Cylindromatosis (CYLD) deubiquitinase to inactivate the tumor suppressor CYLD and to activate the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway in hypoxic conditions.Citation42 E6 interacts with p300/cAMP response element binding protein (CREB)Citation43,Citation44 and interferon regulatory factor 3 (IRF-3)Citation45 to regulate gene expression and with c-Myc to induce upregulation of human telomerase reverse transcriptase to promote cell immortalization.Citation46,Citation47,Citation48 HPV16 E6 in the cytoplasm is also important for the oncogenic activity through its regulation of signal transduction by interactions with cytoplasmic E6BP (Erc55),Citation49 E6TP1,Citation50,Citation51 tumor-necrosis factor (TNF) receptor 1Citation52 and protein tyrosine phosphatase H1.Citation53 In addition to its oncogenic activities, HPV16 E6 also protects HPV16-infected keratinocytes from the innate immune system by suppressing pro-IL-1β expression.Citation54
Figure 1 HPV16 E6 and E7 and alternative RNA splicing. (A) Major functional domains and motifs of the HPV16 E6, E6*I, E6ˆE7 and E7 proteins. HPV16 E6ˆE7 has the N-terminal half of E6 and the C-terminal half of E7. (B) Alternative RNA splicing products of HPV16 E6E7 pre-mRNA. Alternative RNA splicing (dashed lines) takes place from three 5′ ss at nt 191, 221 and 226 to three alternative 3′ ss at nt 409, 526 and 742.Citation27 The majority of RNA splicing occurs from the nt 226 5′ ss to the nt 409 3′ ss to produce E6*I, which is responsible for E7 translation.Citation28 E6*III derived from the nt (226 5′ ss to the nt 3358 3′ ss and E6*IV derived from the nt 226 5′ ss to the nt 2709 3' ssCitation29 are not included in this diagram. (C) Illustration of a ribosomal scanning model in HPV16 E6 and E7 translation, which is regulated by RNA splicing. Full-length E6 is translated from the unspliced E6 mRNA (upper diagram). E7 (nt 562–858) is translated from spliced E6*I mRNA in which a premature stop codon (UAA) is introduced by RNA splicing to form the E6*I ORF (lower diagram), which enlarges the space between the two ORFs. This enables a scanning ribosome to terminate translation of E6*I and reinitiate translation of E7. Nucleotide positions are numbered according to the HPV16 reference genome (PaVE, http://pave.niaid.nih.gov).
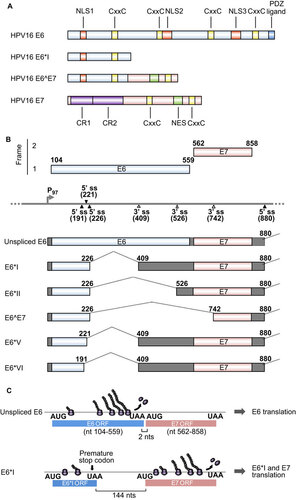
The E7 oncoprotein consists of ∼100 aa. In HPV16 and 45, two CxxC zinc-binding motifs (Figure ) and a hydrophobic surface are essential for E7 homodimerization and protein stability.Citation55 E7 is largely distributed in the nucleus, with a small fraction shuttling to the cytoplasm through two nuclear localization signals and a single nuclear export signal at the C-terminus.Citation56 The N-terminus of E7 contains sequence similarity to a portion of the conserved region 1(CR1) region and the entire CR2 region of adenovirus E1A and the related sequences in SV40 T antigen. Oncogenic E7 binds pRB and the related pocket proteins p107 and p130 with high affinity via an LxCxE motif in CR2 (Figure ). Although low-risk or non-oncogenic E7 only weakly binds pRB, oncogenic E7 induces pRB degradation by interacting with the cullin 2 ubiquitin ligase complex.Citation57 E7 induces aberrant cell cycle progression through upregulation of p21Citation58,Citation59 and p16.Citation60 E7 also induces chromosomal instability and aneuploidy through association with γ-tubulin; this inhibits it from being recruited to the centrosomeCitation61 and complements the requirement for cyclin-dependent kinase 6 (CDK6), ERBB3, FYN, adaptor-associated protein kinase 1 and testis-specific serine kinase 2 for cell survival in colorectal cancer cells.Citation62 Although E7 from both high-risk and low-risk HPVs interacts with p300,Citation63 p300/CBP protein-associated factor (PCAF),Citation64 steroid receptor co-activator 1Citation65 and p600,Citation66 these interactions are not sufficient for E7-mediated transformation.
HPV oncogenes and RNA splicing
The HPV E6 and E7 oncogenes are juxtaposed in two different reading frames in the HPV genomes. Low-risk HPVs and high-risk HPVs utilize different strategies to express E6 and E7. In low-risk HPVs, such as HPV1, 2, 6 and 11, E6 and E7 are transcribed individually from two independent promoters. In contrast, E6 and E7 in high-risk HPVs, such as HPV16, 18, 31, 45 and 58, are transcribed as a single polycistronic E6E7 pre-mRNA from a single early gene promoter upstream of the E6 coding region (such as P97 in HPV16). Although early transcripts of both low-risk and high-risk HPVs contain an intron with alternative splice sites overlapping the E1 or E2 coding regions, and both utilize an early polyadenylation signal downstream of the E5 coding region for RNA polyadenylation, a striking feature of the high-risk HPV E6E7 polycistronic transcript is its unique E6 intron structure and alternative RNA splicing in the E6 coding region.Citation29 The E6 transcript from low-risk HPVs does not have an E6 intron and thus does not undergo RNA splicing in the E6 coding region.Citation67 In general, the E6 intron (also called intron 1) in high-risk E6E7 polycistronic pre-mRNAs features one major 5′ splice site (5′ ss) and one major 3′ splice site (3′ ss), and splicing of this intron disrupts the viral E6 ORF, preventing translation of full-length E6.Citation28,Citation68 Moreover, the E6 intron may extend into the E7, E2 or E4 coding regions through the use of an alternative 3′ ss further downstream of the E6 ORF. In the case of HPV16, alternative E6 intron splicing of the polycistronic E6E7 pre-mRNA leads to the production of seven RNA splicing isoforms, E6*I, E6*II, E6*III, E6*IV, E6*V, E6*VI and E6∧E7 in addition to a full-length, unspliced E6 mRNA (Figure ).Citation27,Citation29
As diagrammed in Figure , the E6 intron of HPV16 bears three alternative 5′ ss in the E6 ORF and three alternative 3′ ss either in the E6 or E7 ORFs. The splicing of intron 1 from HPV16 E6E7 pre-mRNA is highly efficient and depends on intron definition. The majority of the spliced products in cervical cancer and its derived cell lines are E6*I, derived from splicing of the nucleotide (nt) 226 5′ ss to the nt 409 3′ ss. Preferential selection of this pair of splice sites over the other splice sites crossing over the intron minimizes the length of the intron in RNA splicing for energy savingCitation27,Citation69,Citation70 and is dictated by an adenosine at nt 385 within the branch-point sequence AACAAAC in the virus genome.Citation27 HPV16 E6*I encodes a truncated E6*I protein with 43 aa residues, which is in general expressed at levels below the detection threshold. Although the function of the HPV16 E6*I protein remains unknown, HPV18 E6*I appears to play a dominant negative role with regard to full-length E6 oncoproteinCitation71,Citation72,Citation73 and to induce proteasomal degradation of PDZ proteins, including the tumor suppressor hDlg.Citation74 Thus, E6*I splicing is not a viral strategy to produce a potent E6*I protein, but rather a strategy to create an E7 mRNA that can be translated into E7 oncoprotein.Citation28 Because the E6 ORF is only two nucleotides upstream of the E7 ORF in the HPV16 genome, a scanning ribosome is unable to efficiently re-initiate E7 translation from an RNA containing the unspliced, full-length E6 ORF because translation termination of E6 and re-initiation of E7 translation has to occur within the two nucleotides. Subsequently, only the E6 oncoprotein is translatable from the unspliced, intact E6 ORF-containing RNA (Figure ).Citation27,Citation68,Citation75,Citation76 The same is true for translation of HPV18 E6 and E7. However, splicing that produces E6*I creates a premature stop codon immediately downstream of the splice junction and increases the distance between the E6*I ORF and the E7 ORF to >130 nts in both HPV16 and HPV18, a much better condition for scanning ribosomes to re-initiate E7 translation.Citation77 Therefore, the most abundant spliced RNA, E6*I, actually functions as an E7 mRNA for efficient E7 translation (Figure ).Citation27,Citation28,Citation68
In spite of the importance of E6 intron splicing in the production of E7, the regulatory mechanism underlying this splicing event remains largely unexplored. Epidermal growth factor activates extracellular regulated protein kinases 1/2 (Erk1/2), and this activation appears to suppress the HPV16 E6 intron splicing, resulting in increased E6, but decreased E7, translation.Citation76 HPV16 E5 promotes epidermal growth factor receptor activationCitation78,Citation79,Citation80,Citation81 and therefore, may regulate splicing of the E6 intron through activation of the epidermal growth factor pathway. Both HPV16 E2 and E6 act as RNA-binding proteins and suppress splicing of the HPV16 E6 intron,Citation82 suggesting the presence of a positive feedback loop of E6 production.
MERKEL CELL POLYOMAVIRUS
MCV and oncogenes
Polyomaviruses are distantly related to papillomaviruses and were formerly categorized with papillomaviruses into the papovavirus family, a taxonomy which is no longer used. Among the nine species of human polyomaviruses, MCV is the only one proven to be an etiologic agent of human cancer. MCV infection in immune-suppressed individuals can result in MCC, an aggressive form of nonmelanoma skin cancer. MCV has been identified in ∼80% of MCCs.Citation83 Established MCC cell lines contain integrated MCV DNA encoding a mutant T antigen that prevents replication of the integrated virus from being autoactivated.Citation84 This form of mutation in T antigen also affects epitope recognition by cytotoxic T cells.Citation85 Although the other human polyomaviruses, BK polyomaviruse (BKV) and JC polyomaviruse (JCV) , KI polyomaviruse (KIV), WU polyomaviruse (WUV), H polyomaviruse 6 (HPyV6), H polyomaviruse 7 (HPyV7), H polyomaviruse 9 (HPyV9) and trichodysplasia spinulosa-associated polyomaviruses (TSV), are oncogenic in rodents and nonhuman primates, their association with human cancer remains unknown.
Polyomaviruses have been studied for their oncogenic activities since the discovery of SV40.Citation86 SV40 is one of the most common latent viruses in rhesus monkeys and is capable of immortalizing and transforming rodent, but not human, cells. The oncogenicity of SV40 is mediated by large tumor (T) antigen (LT) and small T antigen (sT). ST is a spliced isoform of T antigens. The oncogenic activity of MCV, like that of SV40, is attributed to LT and sT. MCV LT and sT (Figure ) are well conserved with those of SV40. The MCV LT DnaJ domain binds to Hsc70 to promote proper folding of proteins after translation.Citation87 MCV LT and 57kT antigens interact with pRB through their N-terminal LxCxE motif.Citation84 The protein phosphatase 2A (PP2A) binding domain of MCV sT interacts with PP2A, as also seen with SV40 sT,Citation87,Citation88 but mutation of the PP2A binding domain does not impair the oncogenicity of MCV sT.Citation89 MCV LT, but not SV40 LT, interacts with Vam6p.Citation90 Vam6p binds to the N-terminus of MCV LT and changes its subcellular localization from the cytoplasm to the nucleus, but its association with the MCC tumorigenic process remains unclear.
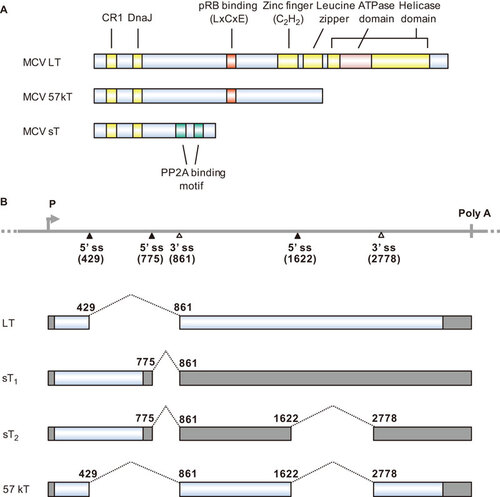
Figure 2 MCV T antigens and alternative RNA splicing. (A) Functional domains and motifs are indicated for the MCV large T antigen (LT), 57 kDa T antigen (57kT) and small T antigen (sT). (B) Scheme of alternative splicing of MCV T antigens. T antigen ORFs created by alternative RNA splicing (dashed lines) are in gray and 5′ and 3′ untranslated regions are in dark gray. Nucleotide positions are numbered according to the MCV genomic sequence (GenBank: NC_010277.1).
MCV oncogenes and RNA splicing
The early pre-mRNAs of MCV undergo alternative RNA splicing to produce four mRNA isoforms: LT, sT1, sT2 and 57kT (Figure ). To date, it remains unknown what RNA cis-element(s) or trans-acting cellular factors are involved in regulating the alternative RNA splicing. LT mRNA encodes LT antigen. Both sT1 and sT2 mRNAs encode sT, but differ in the length of the 3′ untranslated region. The 57kT mRNA corresponds to the 17kT mRNA in SV40 and encodes a 57-kDa protein missing a middle section of LT (Figure ). In contrast to SV40 sT and MCV LT, MCV sT exhibits oncogenic activity: it transforms rodent fibroblasts and promotes serum-independent growth of human fibroblasts.Citation89 This is because MCV sT functions differently from SV40 sT; MCV sT promotes eukaryotic translation initiation factor 4E-binding protein 1 hyperphosphorylation and cap-dependent translation in a PP2A-independent manner,Citation89 whereas SV40 sT induces dephosphorylation of 4E-binding protein 1 in a PP2A-dependent manner and inhibits cap-dependent translation.Citation91 Moreover, MCV sT suppresses the activity of the E3 ubiqitin ligase SCFFbw7, which degrades MCV LT, and stabilizes MCV LT through its LT-stabilization domain;Citation92 SV40 sT does not have an LT-stabilization domain. Although inhibition of SCFFbw7 stabilizes MCV LT, introduction of mutations in the sT LT-stabilization domain decreases LT protein levels and eliminates synergism in MCV DNA replication and sT-induced cell transformation.Citation92 MCV sT is detected in 92% of MCC tissues, whereas MCV LT is only detected in 75% of the tissues.Citation87,Citation89,Citation93 However, the full function of MCV sT in MCC cells requires other T antigens, such as LT and 57kT.Citation85,Citation94
EPSTEIN-BARR VIRUS
EBV and oncogenes
EBV is a human γ-herpesvirus best known as the cause of infectious mononucleosis,Citation95 but it has been also associated with Burkitt lymphoma, nasopharyngeal carcinoma in southeastern Asia, natural killer (NK) cell leukemia, extranodal NK T-cell lymphoma and Hodgkin and non-Hodgkin lymphomas, accounting for ∼1% of cancer cases in humans worldwide.Citation21,Citation96 EBV is widespread in all human populations, with >90% of adults being serologically positive.Citation97 A recent comprehensive analysis of the EBV genome revealed that EBV establishes a latent infection in nearly 100% of infected adults through numerous sites.Citation98 In most cases, EBV infects B lymphocytes. Following EBV infection, B lymphocytes are transformed into lymphoblasts and become immortal. In rare cases, EBV also causes malignancy by infecting T cells and NK cells.Citation99,Citation100,Citation101
Latent membrane protein-1 (LMP1) is considered to be a major viral oncogene of EBV. LMP1 is expressed in EBV-associated lymphoma and is essential for B-cell transformation and disruption of cellular signal transduction.Citation102,Citation103,Citation104 Although EBV nuclear antigen 1 (EBNA1) is one of the earliest viral proteins expressed after infection and is the only latent protein consistently expressed in EBV-associated tumors, EBNA1 is not an oncoprotein.Citation105,Citation106 BamHI-A reading frame-1 is also an early gene, but it is expressed as a latent gene in most nasopharyngeal carcinomas.Citation107 BamHI-A reading frame-1 may play an important role in nasopharyngeal oncogenesis, because it transforms rodent fibroblasts and primary epithelial cells and enhances tumor formation.Citation108,Citation109,Citation110,Citation111 The expression levels of EBV genes depend on the latency status of EBV, which is classified as latency 0, I, II or III. Hodgkin lymphomas are associated with latency II, in which EBNA1 and LMPs are expressed, but Burkitt lymphoma is associated with latency I, in which only EBNA1 is expressed. Nasopharyngeal carcinoma and T-cell and NK lymphomas display EBV latency I/II, an intermediate between latency I and II.
LMP1 is a 62-kDa integral membrane protein and contains 386 aa residues, with a short cytoplasmic N-terminus of 24 aa, six transmembrane domains of 162 aa (from aa 25 to aa 186) and a cytoplasmic C-terminus of 200 aa (Figure ).Citation112,Citation113 LMP1 immortalizes and transforms human B cells, but its oncogenic activity can be interfered by lytic LMP1 (lyLMP1), a truncated form of LMP1 with 258 aa residues expressed in the lytic phase.Citation114 LMP1 drives proliferation of EBV-infected B cells by signaling within the B cells without any ligand for LMP1. The pathway downstream of LMP1 overlaps with that of the CD40 receptor,Citation115 which delivers signaling through TNF receptor associated factors and Janus kinase 3 to activate NF-κB, AP-1, STAT-1, CD83 and CD95. In clonal populations, LMP1 levels vary among cells by more than 100-fold. When expressed at an intermediate level, LMP1 signals through NF-κB to promote cell proliferation, but when expressed at a high level, LMP1 inhibits protein synthesis by activating double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) to induce eukaryotic initiation factor 2α phosphorylation followed by activating transcription factor 4 (ATF4) upregulation. ATF4, in turn, activates the LMP1 promoter.Citation104
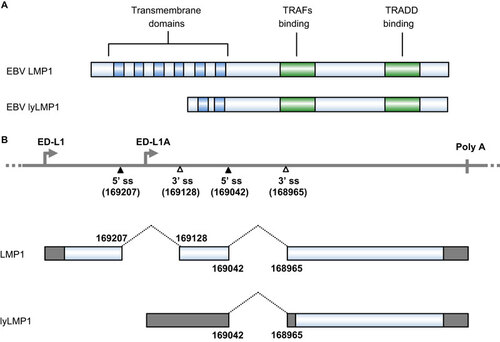
Figure 3 The EBV LMP1 oncoprotein and its lytic variant lyLMP1. (A) Functional domains and motifs are indicated for EBV LMP1 and lyLMP1 in the B95-8 EBV isolate. (B) EBV LMP1 ORF and lyLMP1 are produced by RNA splicing (dashed lines) from two separate transcripts derived either from the ED-L1 promoter or the ED-L1A promoter. Nucleotide positions are numbered according to the genomic DNA sequence of the B95-8 EBV isolate (GenBank: V01555.2). TRAF, TNF receptor-associated factor; TRADD, tumor necrosis factor receptor associated death domain protein.
Although the transmembrane domains of LMP1 activate B-cell apoptosis, the carboxy-terminal domain of LMP1 blocks this effect.Citation116 In general, LMP1 activation leads to overexpression of antiapoptotic molecules, such as B-cell CLL/lymphoma 2 (Bcl-2),Citation117 myeloid cell leukemia 1 (Mcl-1)Citation118 and BCL-2-related protein A1 (Bcl2A1/Bfl-1),Citation116,Citation119 and blocks p53-mediated apoptosis through the induction of anti-apoptotic protein A20,Citation120 unfolded protein response (UPR)-induced apoptosis in B cellsCitation116 and ubiquitin C-terminal hydrolase L1 (UCH-L1) with oncogenic properties.Citation121 Complementary to its proliferative function, LMP1 inhibits proapoptotic factors such as BaxCitation122 and induces autocrine factors, such as chemokine (C–C motif) ligand 3 (CCL3) and chemokine (C–C motif) ligand 4 (CCL4), to promote cell proliferation.Citation123 LMP1 also enhances cancer cell motility by upregulating TNF α-induced protein 2 (TNFAIP2) through NF-κB activation.Citation124 Moreover, LMP1 regulates miRNAs to exert both positive and negative roles on cell proliferation. LMP1 upregulates miR-29b to suppress TCL1 oncogene expression,Citation125 but downregulates miR-203 to increase E2F transcription factor 3 (E2F3) and cyclin G1 expressionCitation126 and miR-15a to promote MYB and cyclin D1 expression.Citation127
EBV LMP1 and RNA splicing
The LMP1 gene is transcribed from either the ED-L1 or ED-L1A promoter within the BamHI-N region of the EBV genomeCitation112,Citation128 and produces two LMP1 isoforms from the two alternative promoters (Figure ). Double splicing of ED-L1 pre-mRNA produces the LMP1 ORF encoding 386 aa residues, whereas single splicing of ED-L1A pre-mRNA produces the lyLMP1 ORF encoding 258 aa residues starting from the 129th methionine of LMP1 in B95-8 EBV isolates.Citation129,Citation130 Thus, lyLMP1 differs from LMP1 by lacking the N-terminal 129 aa residues of LMP1. However, the lyLMP1 transcript is non-coding in most EBV isolatesCitation131 and is upregulated during the lytic phase of EBV replication following activation of the lytic promoter ED-L1A.Citation132,Citation133 In contrast to LMP1, lyLMP1 has no transforming activity in rodent cells, nor does it alter the phenotypes of human B lymphocytes.Citation134 The biological activity of lyLMP1 appears to be the negative regulation of LMP1-signaling pathways and LMP1-mediated oncogenesis.Citation114,Citation135
What regulates splicing of LMP1 RNA remains largely unexplored. EBV EB2 (also called SM) functions as a trans-acting factorCitation136,Citation137 in regulating RNA splicing via its interaction with the cellular oncogenic splicing factor SRSF3 (serine/arginine-rich splicing factor 3).Citation138 Although EBV EB2 is not expressed in the latent stage of EBV infection, increased SRSF3 expression in lymphoma might play a role in LMP1 splicing and is worth investigating.
HUMAN T-CELL LEUKEMIA VIRUS 1
HTLV-1 and oncogenes
HTLV-1 is a delta retrovirus and is associated with ATL. Both HTLV-1 and ATL are endemic within the southwestern part of Japan, the Caribbean basin, and South Africa. Approximately 1%–5% of individuals with HTLV-1 infection develop ATL after 20–30 years of latency.Citation139,Citation140 Discovery of HTLV-1 occurred independently in the United States and Japan in the late 1970s. A retrovirus with type-C morphology was isolated from the blood of an African-American patient with cutaneous T-cell lymphoma and named human cutaneous T-cell lymphoma virus.Citation13 In parallel, adult T-cell leukemia was found in the southwestern part of Japan,Citation141,Citation142 and a retrovirus with type-C morphology was also isolated from T cells from those patients and named adult T-cell leukemia virus.Citation14,Citation15,Citation143,Citation144 The two viruses were later found to be identical, and are now referred to as HTLV-1. Three related viruses, HTLV-2, -3 and -4, have also been reported. HTLV-2, which was isolated from a hairy T-cell leukemia, immortalizes human T cells in vitro,Citation145 although its association with human disease has not yet been established. HTLV-3 and -4 were detected from their genome sequences, but their relation to human disease also remains unknown.Citation146,Citation147
Tax, a viral oncogene of HTLV-1, encodes a 40-kDa nuclear phosphoprotein consisting of 353 aa residues and confers the transforming properties of HTLV-1. Tax immortalizes T lymphocytes and induces leukemia in transgenic mice.Citation148,Citation149 It promotes viral transcription from a promoter located within the long terminal repeat (LTR).Citation150,Citation151,Citation152 The N-terminus of Tax (Figure ) directly binds to CREB to form a ternary complex of TRE–Tax–CREB (Tax-responsive element–Tax–CREB) within the viral promoter.Citation154,Citation155 Binding of Tax to CREB enhances CREB homodimerization, which strengthens its association with promoter DNA.Citation156 TRE activation requires Tax homodimerization, which involves CREB homodimerization. Tax also recruits the transcriptional co-activators CREB-binding protein (CBP) and p300Citation157,Citation158,Citation159 and PCAF.Citation160,Citation161 Tax transactivates specific cellular transcripts by interacting with DNA-binding serum responsive factor, which recruits Tax to specific cellular promoters such as those of c-FOS, EGR-1 and EGR-2. Citation162,Citation163,Citation164 Tax activates the NF-κB pathwayCitation165 to enhance the expression of IL-2,Citation166,Citation167 IL-2R,Citation168 IL-15Citation169 and IL-15R,Citation170 leading to the formation of an autocrine loop for the proliferation of HTLV-1-infected T cells. Tax inactivates the tumor suppressor p53Citation171 and its homologues p73α and β,Citation172,Citation173 but activates canonical Wnt signaling in the presence of a Wnt pathway-associated protein, disheveled-associating protein with a high frequency of leucine residues (DAPLE).Citation174 Tax binds INT6/EIF3E, a subunit of the translation initiation factor eukaryotic initiation factor 3, and upstream frameshift protein 1 (UPF1) to inhibit nonsense-mediated mRNA decay.Citation175
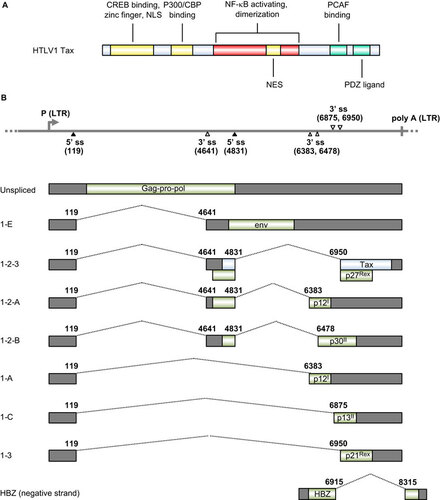
Figure 4 HTLV-1 viral RNA splicing and Tax oncogene expression. (A) Functional domains and motifs are indicated for the HTLV-1 Tax oncoprotein. (B) Alternative RNA splicing products of HTLV-1 viral genes. Seven positive-strand transcripts (1-E, 1-2-3, 1-2-A, 1-A, 1-2-B, 1-C and 1-3) and one negative strand transcript (HBZ) are produced by alternative RNA splicing of the transcripts derived from either a 5′ or 3′ long terminal repeat (LTR). The HTLV-1 Tax ORF in blue is present in the 1–2–3 mRNA, which is spliced from the nt 119 5′ ss to the nt 4641 3′ ss and then from the nt 4831 5′ ss to the nt 6950 3′ ss. Nucleotide positions are numbered according to the HTLV-1 genomic RNA sequence, starting from the first nucleotide in the pre-mRNA.Citation153
Despite its oncogenic activity, Tax expression is low or undetectable in tumor cells from adult T-cell leukemia.Citation176,Citation177,Citation178 The current consensus is that Tax is necessary for initiating cell transformation, but in later stages, acquired genetic and epigenetic changes and alternative growth-promoting pathways replace the roles of Tax to maintain adult T-cell leukemia when Tax is no longer expressed.
HTLV-1 RNA splicing and production of tax mRNA
Reverse transcription of HTLV-1 genomic RNA into DNA and subsequent DNA integration into the host cell genome is a necessary step for retroviral replication. All HTLV-1 genes, except the newly described HTLV-1 basic leucine zipper factor (HBZ) gene, are transcribed from a single promoter within the 5′ LTR as a single polycistronic pre-mRNA transcript that is polyadenylated by using a polyadenylation signal in the 3′ LTR.Citation67 HBZ is encoded by an antisense transcript derived from a minor promoter in the 3' LTR of the HTLV-1 genome.Citation179 Both the major polycistronic transcript and the minor monocistronic antisense transcript contain introns and are subject to regulation by RNA splicing.Citation180 The major polycistronic pre-mRNA transcript covers almost the entire viral genome. Gag-Pro-Pol, a precursor polyprotein, is translated from the unspliced form of the polycistronic RNA. However, the RNA also has two introns and three exons with two alternative 5′ ss at nt 119 and nt 4831, and five alternative 3′ ss at nts 4641, 6383, 6478, 6875 and 6950. The HBZ monocistronic pre-mRNA has only one intron, and the antisense HBZ transcript undergoes RNA splicing from nt 8315 to 6915.Citation179 This leads to extensive alternative RNA splicing and produce nine types of spliced and unspliced mRNAs (Figure ). The Tax ORF is produced by double RNA splicing of the polycistronic RNA, with the first intron splice from nt 119 to 4641 and the second intron splice from nt 4831 to 6950 in the spliced 1–2–3 form of the RNA (Figure ). What controls or regulates the alternative RNA splicing in HTLV-1 infection and gene expression remains to be investigated. RNA cis-elements and trans-acting factors responsible for HTLV-1 alternative RNA splicing remain unknown.
KAPOSI'S SARCOMA-ASSOCIATED HERPESVIRUS
KSHV and viral genes with oncogenic activities
KSHV or human herpesvirus 8 was discovered in 1994 as a member of the human gamma herpesvirus family, joining EBV.Citation20,Citation181 Infection of immune-compromised individuals with KSHV has been associated with the development of endothelial cell-derived Kaposi's sarcoma and at least two B cell lymphoproliferative diseases: primary effusion lymphoma and multicentric Castleman's disease.Citation21 However, studying KSHV pathogenesis and oncogenesis has been hindered by lack of a meaningful animal model and susceptible cell culture, although primary endothelial cells provide limited KSHV replication Citation182,Citation183. Two immortalized cell lines, KS Y-1 and SLK, were once used for KS and KSHV studies,Citation184,Citation185 but the KS Y-1 cell line is cross-contaminated with the T24 urinary bladder cancer cell line (ATCC HTB-4) and SLK is a contaminant of a known renal carcinoma cell line, Caki-1.Citation186 Thus, neither are of endothelial origin. Primary rat embryonic metanephric mesenchymal precursor cells are susceptible to KSHV infection and transformation, but only shed a limited number of infectious virions.Citation187 Primary effusion lymphoma-derived B-cell lines are commonly infected with KSHV at the latent stage and can be induced to produce low levels of KSHV virions,Citation188,Citation189 but primary B lymphocytes from peripheral blood or tonsillar tissue are refractory to infection by KSHV,Citation190,Citation191 and their limited infection may require cocultivation with KSHV-positive cells.Citation185 The human MC116 cell line, which has characteristics of transitional B cells,Citation192 can be infected with limitation by the KSHV.219 virus, which also infects Vero cells and produces high yields of virus.Citation193 Humanized bone marrow, liver and thymus (BLT) mice infected by inoculation with KSHV.219 virus via the oral and vaginal routes could be a useful model for studying the pathogenesis and transmission of KSHV.Citation194
KSHV encodes several important proteins that have some oncogenic activity for inducing cell proliferation, immortalization, transformation and signaling; cytokine production; immune evasion; antiapoptosis activity; and angiogenesis. These include the viral latent proteins latent-associated nuclear antigen (LANA), vFLIP (a FADD (Fas-associated protein with death domain)-like interferon converting enzyme or caspase 8 (FLICE) inhibitory protein), and vCyclin and the viral lytic proteins G-protein coupled receptor (vGPCR) interferon regulatory factor 1 (vIRF-1) and K1. Although the true oncogenic nature of each protein remains to be defined, accumulating evidence indicates that each of them contributes some aspect to KSHV oncogenesis. Thus, a full spectrum of KSHV-induced malignancy might require multiple oncogenic products to work together in the presence of host and environmental cofactors. For example, both LANA and vIRF-1 target the cellular tumor suppressor p53.Citation195,Citation196 LANA also inhibits pRB and PP2A.Citation197 vCyclin, an activator of CDK4/6,Citation198 downregulates p27kip1, a CDK inhibitor,Citation199 and counters the senescence/G1 arrest response that results from NF-κB hyperactivation.Citation200 Both vFLIP and K1 activate the NF-κB signal pathway to prevent B cell apoptosis.Citation201,Citation202 vGPCR and K1 affect the AKT and NF-κB signal pathwaysCitation203,Citation204,Citation205 and contribute to angioproliferative and inflammatory Kaposi's sarcoma lesions.Citation206,Citation207 More importantly, the latent locus of the KSHV genome by itself shows B cell oncogenicity in transgenic mice.Citation208
Transcription and RNA splicing in the KSHV latent locus
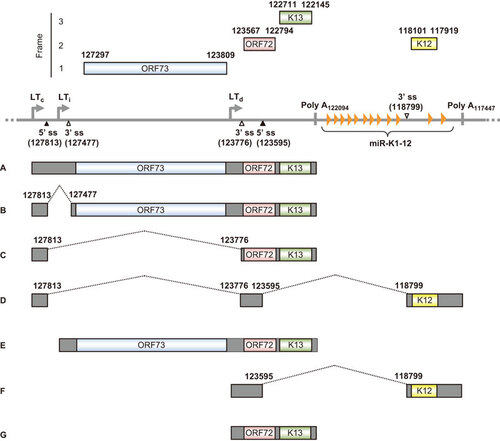
The KSHV latent locus encodes multiple viral latent genes, including LANA (ORF73), vCyclin (ORF72), vFLIP (ORF71 or K13), all viral miRNAs and kaposin (K12). Expression of these latent genes from this locus is driven by three promoters (Figure ): two constitutive latent promoters (LTc and LTd) and an inducible latent promoter (LTi) that is inactive but can be induced by the lytic switch protein RTA encoded by ORF 50.Citation209 Although LTd is constitutively active, mirroring LTc, its transcriptional activity can be further boosted by expression of RTA, reminiscent of LTi.Citation209 As diagrammed in Figure , both the LTc and LTi transcripts are tricistronic and are polyadenylated by using a common poly A (pA) signal at nt 122094 in the virus genome. The LTc transcript has an intron with two alternative 3′ ss and is thus subject to regulation by alternative RNA splicing, whereas the LTi transcript does not have an intron and is not spliced. Consequently, LANA is expressed from the unspliced RNA species A or E or the spliced RNA species B by using a proximal 3′ ss that preserves the intact ORF73 ORF. In KSHV-infected cells, the majority of LANA is translated from abundant A RNA. Viral vCyclin and vFLIP are expressed mainly from spliced C RNA derived by selection of a distal 3′ ss for RNA splicing, with vFLIP being expressed from this transcript by using an internal ribosome entry site residing in the vCylcin ORF.Citation210 Occasionally, an LTc transcript is double spliced to the K12 ORF region by using another pA signal further downstream at nt 117432 (RNA species D).Citation211 This last splicing strategy presumably enables the LTc transcript to encode K12. The LTd transcript can be spliced to the K12 ORF for polyadenylation to encode K12, or it can remain unspliced, in which case it is polyadenylated by using the same pA signal as the LTi RNA E to encode vCyclin and vFLIP. The intron from the LTd RNA species F could be a source of KSHV miRNAs.Citation209,Citation212 However, very little is known about the splicing regulation of these latent transcripts.
Figure 5 Transcription map of KSHV ORF73 (LANA), ORF72 (v-Cyclin), ORF71 (K13, v-FLIP) and K12 (kaposin). The diagram shows major alternative splicing products of KSHV ORF73, ORF72, ORF71 and K12. The heavy line indicates the corresponding latent locus region of KSHV genome. Three promoters (LTc, constitutive promoter; LTi, RTA-inducible promoter; LTd, downstream promoter), two polyadenylation signals (poly A at nt 122094 and nt 117432) and alternative 5′ ss and 3′ ss in this region are indicated. Twelve viral miRNAs (miR-K1-12) clustering downstream of the K13 ORF are shown as orange triangles. Boxes above the line represent ORFs for ORF73, ORF72, K13 and K12. Below the line are common RNA species (A–G) derived from alternative RNA splicing from this region. Nucleotide positions are indicated according to KSHV genome (GenBank: U75698.1).Citation181
HEPATITIS B VIRUS AND HEPATITIS C VIRUS
HBV and hepatocellular carcinoma
More than 50% of liver cancers worldwide are attributed to HBV infection. HBV is a DNA virus whose ∼3.2 kb, partially double-stranded, circular DNA genome is covered by a nucleocapsid core and an outer lipid envelope. The HBV envelope consists of the small (S), middle (M) and large (L) envelope proteins, which are multiple-transmembrane proteins that share the same C-terminal domain (corresponding to the S protein) but which differ at their N-terminal domains. The HBV genome encodes four viral genes: C (HBcAg), X (HBx), P (DNA polymerase) and S (HBsAg). The S gene encodes a long ORF with three in-frame ‘start’ (ATG) codons that divide the gene into three sections, pre-S1, pre-S2 and S. HBV enters susceptible liver cells when the receptor-binding region of pre-S1 specifically interacts with the functional cellular receptor NTCP (sodium taurocholate cotransporting polypeptide), a multiple transmembrane transporter predominantly expressed in the liver; this results in liver infection and virus replication.Citation213 Persistence of chronic HBV infection for decades is linked to liver cirrhosis and the development of hepatocellular carcinoma (HCC). Although HBV infection is prevalent worldwide, chronic HBV infection is most common in East Asia through perinatal transmission and in Africa via childhood infection.Citation214,Citation215,Citation216 Continuous cycling of immune clearance and regeneration of hepatocytes during chronic HBV infection is considered a risk factor for HCC development. HBx is a transactivating protein that interacts with protein arginine methyltransferase 1 to regulate the expression of cellular genes,Citation217 stimulating cell growth-promoting genes and inactivating growth regulating molecules. HBx regulates cell cycle progression and cell proliferation by activating NF-κB;Citation218 upregulating vascular endothelial growth factor receptor 3 in hepatocarcinogenesis;Citation219 stimulating cell migration, growth in soft agar, and spheroid formation; and promoting ‘stemness’ in the pathogenesis of HCC.Citation220 HBx and aflatoxin B1 synergistically cause hepatitis, steatosis, and liver hyperplasia in transgenic zebrafish.Citation221
HBV RNA splicing and HCC
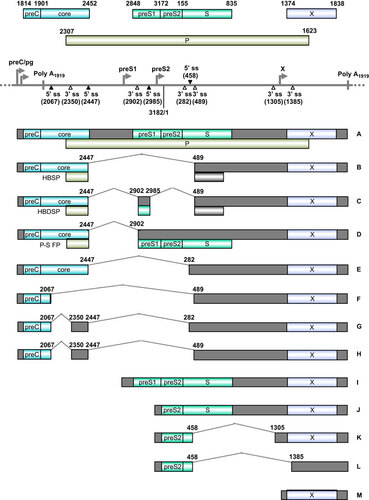
Transcription of HBV RNAs is controlled by four different promoters: preCore/pre-genomic (preC/pg, for the 3.5-kb core RNA that codes for viral core protein and DNA polymerase), pre-S1 (for the 2.4-kb pre-S1 RNA that encodes pre-S1), major pre-S2/S (for the 2.1-kb pre-S2/S RNAs that encode the pre-S2 and S proteins) and X (for the 0.7-kb RNA encoding HBx antigen) (Figure ).Citation223,Citation224 However, all HBV RNA transcripts are polyadenylated by using a single pA signal downstream of the HBx termination codon. With such a genome structure, HBV encodes specific sequence elements to promote extensive splicing of HBV pre-genomic/preC (pg/preC), pre-S1 and pre-S2/S RNAs, a common event during chronic infection, leading to the production of seven additional common splicing variants of pg/preC RNAs and two additional splicing variants of pre-S2/S RNAs (Figure ).Citation225,Citation226 A 2.2-kb singly spliced (spliced from nt 2447–489, Figure RNA B) or doubly spliced (spliced from nt 2447 to nt 2902 and then from nt 2985 to nt 489, Figure RNA C) HBV pg/preC RNA is most common in cultured cells. It appears that this single or double splicing event is regulated by two RNA cis-elements, the post-transcriptional regulatory element at nts 1217–1582Citation225,Citation227 and the intronic splicing silencer (ISS) at nts 2591–3163,Citation228 and by PTB-associated splicing factor in interaction with the post-transcriptional regulatory element.Citation229 The singly spliced RNA encodes an hepatitis B splice-generated protein (HBSP), which contains a small portion of the N-terminal viral polymerase fused with a new ORF produced by RNA splicing.Citation230 HBSP is associated with pathogenesis after HBV infection and increases the risk of development of HCC by promoting viral replication and protein productionCitation231 and liver fibrosis.Citation232 HBSP expression activates the HBV S1 promoter, S2 promoter, enhancer I and core upstream regulatory sequences.Citation233 HBSP interacts with cathepsin B to enhance hepatoma cell migration and invasionCitation234 and induces T-cell responses in human leukocyte antigen-transgenic mice and HBV-infected patients.Citation235 The doubly spliced RNA encodes a ∼15-kDa hepatitis B doubly spliced protein which may play a role in viral transcription and DNA replication.Citation233,Citation236 Polymerase-surface fusion protein (P-S FP), which is encoded by another spliced transcript derived by splicing from nt 2474 to 2902 of pg/preC RNA (Figure , RNA D), might regulate HBV replication.Citation237,Citation238 The functions of the spliced pre-S2/S RNAs remain unknown, and the HBx transcript does not undergo RNA splicing.
Figure 6 Genome structure and transcription map of HBV. The full-length, circular HBV genome (GenBank: X02496.1)Citation222 is illustrated in a linear form for better presentation of tail-to-head (3182/1) junction, four promoters (preC/pg, preS1, preS2 and X), a single poly A site at nt 1919, four alternative 5′ ss (filled triangles) and six alternative 3′ ss (open triangles). Above the linear HBV genome are viral ORFs, each with the numbered positions of the first nt of the start codon (including the in-frame start codon) and the last nt of the stop codon. Below the linear HBV genome are the RNA species (A–M) commonly derived from alternative RNA splicing, with the coding exons in colored boxes and the non-coding exons in grey boxes. The dotted lines indicate the introns and splicing directions for each RNA species, with the mapped splice site positions being numbered by nt positions in the HBV genome. HBSP, HBDSP and P-S FP are the ORFs created by alternative splicing of the preC/pg RNA. HBSP, hepatitis B splice-generated protein; HBDSP, hepatitis B doubly spliced protein; P-S FP, polymerase-surface fusion protein.
HCV and oncogenesis
HCV is a positive-strand RNA virus with a ∼9.6-kb genome. The HCV genome encodes a single ORF for a polyprotein of about 3000 aa residues, which is cleaved into 10 different viral proteins. In order from the N-terminus to C-terminus, these are a viral core protein, the two envelope proteins E1 and E2, and seven non-structural proteins p7 (NS1)–NS2–NS3 (protease/RNA helicase)–NS4A–NS4B–NS5A–NS5B (RNA polymerase). The HCV lifecycle is restricted to the cytoplasm and is not regulated by nuclear RNA splicing. Accumulation of HCV RNA in the infected cells requires an interaction of host miR-122 with the 5′-non-coding region of HCV RNA.Citation239,Citation240 HCV grows in Huh-7-derived cell lines.Citation241,Citation242,Citation243 Similar to HBV, chronic HCV infection over decades is strongly associated with liver cirrhosis and HCC, and chronic inflammation and regeneration of hepatocytes through the immune response are prognostic risk factors. At least four of the HCV gene products, the viral core, NS3, NS4B and NS5A, exhibit transformation potential in tissue culture, and several oncogenic pathways can be altered by the expression of these HCV proteins, thus presumably contributing to HCC development.Citation244,Citation245,Citation246 Today, approximately ∼94% of chronic HCV genotype 1 infection in adults can be cured in 8 weeks by a once-daily fixed-dose combination of the NS5A inhibitor ledipasvir 90 mg and the nucleotide analog polymerase (NS5B) inhibitor sofosbuvir 400 mg.Citation247,Citation248,Citation249
REMARKS
The discoveries in the past two decades of KSHV and MCV, the introduction of HPV vaccines for the prevention of cervical cancer and the successful growth of HCV in Huh-7-derived cell lines and treatment of HCV chronic infection with sofosbuvir/ledipasvir are historical landmarks in tumor virology and human cancer research. These remarkable successes reiterate that the global fight against human cancers will continue to receive great support from our tremendous efforts in searching for new tumor-causing viruses and in understanding the basic biology of tumor viruses. Oncogenes, signal transduction and RNA splicing were all discovered by tumor virologists in the late 1970s and early 1980s and have had tremendous impact on today's cancer research portfolio around the world. As discussed in this review, all tumor viruses express oncogenes (HPV, EBV, MCV and HTLV-1) or genes with oncogenic activities (KSHV, HBV and HCV) that immortalize and transform host cells. However, expression of these defined oncogenes, although regulated at the transcriptional level, is also profoundly under the control of alternative RNA splicing at the post-transcriptional level, and to date we know only a little about the mechanisms that regulate RNA splicing in the context of viral infection and viral oncogenesis of host cells. Exploring RNA cis-elements and cellular trans-acting splicing factors in the regulation of alternative RNA splicing of tumor viruses has shed some light on the mechanisms over the past 18 years and will most likely continue to be a prospective focus in viral oncogene research. Gaining further understanding of how the cell- or tissue-specific expression of splicing factors is involved in splicing of viral oncogene RNA will be a formidable challenge, but will provide some fundamental insight into how alternative RNA splicing and viral gene expression take place in a cell- or tissue-specific manner. Looking back on 100 years of tumor virology history, we have learned a great deal about human carcinogenesis from many landmark discoveries in tumor virology. In looking forward into the twenty-first century, understanding and manipulation of RNA splicing in the development and control of human cancers will definitely trigger another wave of discoveries in RNA biology. Human tumor viruses will inevitably be windows into this complex nature.
This research program was supported by the Intramural Research Program of the National Cancer Institute, Center for Cancer Research, National Institutes of Health. Masahiko Ajiro was supported by the Japan Society for the Promotion of Science (JSPS) Research Fellowship for Japanese biomedical and behavioral researchers at the National Institutes of Health.
- de Martel C, Ferlay J, Franceschi S et al.Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol2012;13: 607–615.
- Rous P.A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med1911;13: 397–411.
- Shope RE, Hurst EW.Infectious papillomatosis of rabbits: with a not on the histopathology. J Exp Med1933;58: 607–624.
- Bittner JJ.Some possible effects of nursing on the mammary gland tumor incidence in mice. Science1936;84: 162.
- Rowe WP, Huebner RJ, Gilmore LK, Parrott RH, Ward TG.Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc Soc Exp Biol Med1953;84: 570–573.
- Hilleman MR, Werner JH.Recovery of new agent from patients with acute respiratory illness. Proc Soc Exp Biol Med1954;85: 183–188.
- Sweet BH, Hilleman MR.The vacuolating virus, S.V. 40. Proc Soc Exp Biol Med1960;105: 420–427.
- Stehelin D, Varmus HE, Bishop JM, Vogt PK.DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature1976;260: 170–173.
- Lane DP, Crawford LV.T antigen is bound to a host protein in SV40-transformed cells. Nature1979;278: 261–263.
- Linzer DI, Levine AJ.Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell1979;17: 43–52.
- Kovesdi I, Reichel R, Nevins JR.Identification of a cellular transcription factor involved in E1A trans-activation. Cell1986;45: 219–228.
- Epstein MA, Henle G, Achong BG, Barr YM.Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt's lymphoma. J Exp Med1965;121: 761–770.
- Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC.Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA1980;77: 7415–7419.
- Hinuma Y, Nagata K, Hanaoka M et al.Adult T-cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc Natl Acad Sci USA1981;78: 6476–6480.
- Miyoshi I, Kubonishi I, Yoshimoto S et al.Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature1981;294: 770–771.
- zur Hausen H.Condylomata acuminata and human genital cancer. Cancer Res1976;36: 794.
- zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW.Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer1974;13: 650–656.
- Durst M, Gissmann L, Ikenberg H, zur Hausen H.A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci USA1983;80: 3812–3815.
- Boshart M, Gissmann L, Ikenberg H, Kleinheinz A, Scheurlen W, zur Hausen H.A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J1984;3: 1151–1157.
- Chang Y, Cesarman E, Pessin MS et al.Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science1994;266: 1865–1869.
- Cesarman E.Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol2014;9: 349–372.
- Feng H, Shuda M, Chang Y, Moore PS.Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science2008;319: 1096–1100.
- Howley PM, Lowy DR.Papillomaviruses.In: Knipe DM, Howley PM (ed.) Fields virology.5th ed.Philadelphia, PA: Lippincott Williams & Wilkins, 2007: 2299–2354.
- Walboomers JM, Jacobs MV, Manos MM et al.Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol1999;189: 12–19.
- Forman D, de Martel C, Lacey CJ et al.Global burden of human papillomavirus and related diseases. Vaccine2012;30: F12–F23.
- Munoz N, Bosch FX, de Sanjose S et al.Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med2003;348: 518–527.
- Ajiro M, Jia R, Zhang L, Liu X, Zheng ZM.Intron definition and a branch site adenosine at nt 385 control RNA splicing of HPV16 E6*I and E7 expression. PLoS ONE2012;7: e46412.
- Tang S, Tao M, McCoy JP Jr, Zheng ZM.The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol2006;80: 4249–4263.
- Zheng ZM, Baker CC.Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci2006;11: 2286–2302.
- Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M.Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA1997;94: 11612–11616.
- Lee SS, Weiss RS, Javier RT.Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA1997;94: 6670–6675.
- Thomas M, Dasgupta J, Zhang Y, Chen X, Banks L.Analysis of specificity determinants in the interactions of different HPV E6 proteins with their PDZ domain-containing substrates. Virology2008;376: 371–378.
- Zhang Y, Dasgupta J, Ma RZ, Banks L, Thomas M, Chen XS.Structures of a human papillomavirus (HPV) E6 polypeptide bound to MAGUK proteins: mechanisms of targeting tumor suppressors by a high-risk HPV oncoprotein. J Virol2007;81: 3618–3626.
- Tao M, Kruhlak M, Xia S, Androphy E, Zheng ZM.Signals that dictate nuclear localization of human papillomavirus type 16 oncoprotein E6 in living cells. J Virol2003;77: 13232–13247.
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM.The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell1990;63: 1129–1136.
- Ansari T, Brimer N, vande Pol SB.Peptide interactions stabilize and restructure human papillomavirus type 16 E6 to interact with p53. J Virol2012;86: 11386–11391.
- Huibregtse JM, Scheffner M, Howley PM.Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol Cell Biol1993;13: 4918–4927.
- Zanier K, Charbonnier S, Sidi AO et al.Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science2013;339: 694–698.
- Zanier K, ould M'hamed ould SA, Boulade-Ladame C et al.Solution structure analysis of the HPV16 E6 oncoprotein reveals a self-association mechanism required for E6-mediated degradation of p53. Structure2012;20: 604–617.
- Nomine Y, Masson M, Charbonnier S et al.Structural and functional analysis of E6 oncoprotein: insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol Cell2006;21: 665–678.
- Ristriani T, Fournane S, Orfanoudakis G, Trave G, Masson M.A single-codon mutation converts HPV16 E6 oncoprotein into a potential tumor suppressor, which induces p53-dependent senescence of HPV-positive HeLa cervical cancer cells. Oncogene2009;28: 762–772.
- An J, Mo D, Liu H et al.Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell2008;14: 394–407.
- Patel D, Huang SM, Baglia LA, McCance DJ.The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J1999;18: 5061–5072.
- Zimmermann H, Degenkolbe R, Bernard HU, O'Connor MJ.The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J Virol1999;73: 6209–6219.
- Ronco LV, Karpova AY, Vidal M, Howley PM.Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev1998;12: 2061–2072.
- Gross-Mesilaty S, Reinstein E, Bercovich B et al.Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc Natl Acad Sci USA1998;95: 8058–8063.
- Veldman T, Liu X, Yuan H, Schlegel R.Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci USA2003;100: 8211–8216.
- Miller J, Dakic A, Chen R et al.HPV16 E7 protein and hTERT proteins defective for telomere maintenance cooperate to immortalize human keratinocytes. PLoS Pathog2013;9: e1003284.
- Chen JJ, Reid CE, Band V, Androphy EJ.Interaction of papillomavirus E6 oncoproteins with a putative calcium- binding protein. Science1995;269: 529–531.
- Gao Q, Srinivasan S, Boyer SN, Wazer DE, Band V.The E6 oncoproteins of high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it for degradation. Mol Cell Biol1999;19: 733–744.
- Singh L, Gao Q, Kumar A et al.The high-risk human papillomavirus type 16 E6 counters the GAP function of E6TP1 toward small Rap G proteins. J Virol2003;77: 1614–1620.
- Filippova M, Song H, Connolly JL, Dermody TS, Duerksen-Hughes PJ.The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J Biol Chem2002;277: 21730–21739.
- Topffer S, Muller-Schiffmann A, Matentzoglu K, Scheffner M, Steger G.Protein tyrosine phosphatase H1 is a target of the E6 oncoprotein of high-risk genital human papillomaviruses. J Gen Virol2007;88: 2956–2965.
- Niebler M, Qian X, Hofler D et al.Post-translational control of IL-1beta via the human papillomavirus type 16 E6 oncoprotein: a novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog2013;9: e1003536.
- Todorovic B, Massimi P, Hung K, Shaw GS, Banks L, Mymryk JS.Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J Virol2011;85: 10048–10057.
- Knapp AA, McManus PM, Bockstall K, Moroianu J.Identification of the nuclear localization and export signals of high risk HPV16 E7 oncoprotein. Virology2009;383: 60–68.
- Huh K, Zhou X, Hayakawa H et al.Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol2007;81: 9737–9747.
- Jian Y, Schmidt-Grimminger DC, Chien WM, Wu X, Broker TR, Chow LT.Post-transcriptional induction of p21cip1 protein by human papillomavirus E7 inhibits unscheduled DNA synthesis reactivated in differentiated keratinocytes. Oncogene1998;17: 2027–2038.
- Noya F, Chien WM, Broker TR, Chow LT.p21cip1 degradation in differentiated keratinocytes is abrogated by costabilization with cyclin E induced by human papillomavirus E7. J Virol2001;75: 6121–6134.
- Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y.pRB family proteins are required for H3K27 trimethylation and polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev2007;21: 49–54.
- Nguyen CL, Eichwald C, Nibert ML, Munger K.Human papillomavirus type 16 E7 oncoprotein associates with the centrosomal component gamma-tubulin. J Virol2007;81: 13533–13543.
- Baldwin A, Li W, Grace M et al.Kinase requirements in human cells: II. Genetic interaction screens identify kinase requirements following HPV16 E7 expression in cancer cells. Proc Natl Acad Sci USA2008;105: 16478–16483.
- Bernat A, Avvakumov N, Mymryk JS, Banks L.Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene2003;22: 7871–7881.
- Avvakumov N, Torchia J, Mymryk JS.Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene2003;22: 3833–3841.
- Baldwin A, Huh KW, Munger K.Human papillomavirus E7 oncoprotein dysregulates steroid receptor coactivator 1 localization and function. J Virol2006;80: 6669–6677.
- DeMasi J, Huh KW, Nakatani Y, Munger K, Howley PM.Bovine papillomavirus E7 transformation function correlates with cellular p600 protein binding. Proc Natl Acad Sci USA2005;102: 11486–11491.
- Zheng ZM.Viral oncogenes, noncoding RNAs, and RNA splicing in human tumor viruses. Int J Biol Sci2010;6: 730–755.
- Zheng ZM, Tao M, Yamanegi K, Bodaghi S, Xiao W.Splicing of a cap-proximal human papillomavirus 16 E6E7 intron promotes E7 expression, but can be restrained by distance of the intron from its RNA 5′ cap. J Mol Biol2004;337: 1091–1108.
- Romfo CM, Alvarez CJ, van Heeckeren WJ, Webb CJ, Wise JA.Evidence for splice site pairing via intron definition in Schizosaccharomyces pombe. Mol Cell Biol2000;20: 7955–7970.
- Shao W, Kim HS, Cao Y, Xu YZ, Query CC.A U1–U2 snRNP interaction network during intron definition. Mol Cell Biol2012;32: 470–478.
- Pim D, Massimi P, Banks L.Alternatively spliced HPV-18 E6* protein inhibits E6 mediated degradation of p53 and suppresses transformed cell growth. Oncogene1997;15: 257–264.
- Pim D, Banks L.HPV-18 E6*I protein modulates the E6-directed degradation of p53 by binding to full-length HPV-18 E6. Oncogene1999;18: 7403–7408.
- Guccione E, Pim D, Banks L.HPV-18 E6*I modulates HPV-18 full-length E6 functions in a cell cycle dependent manner. Int J Cancer2004;110: 928–933.
- Pim D, Tomaic V, Banks L.The human papillomavirus (HPV) E6* proteins from high-risk, mucosal HPVs can direct degradation of cellular proteins in the absence of full-length E6 protein. J Virol2009;83: 9863–9874.
- Tang S, Tao M, McCoy JP, Zheng ZM.Short-term induction and long-term suppression of HPV16 oncogene silencing by RNA interference in cervical cancer cells. Oncogene2006;25: 2094–2104.
- Rosenberger S, de Castro AJ, Langbein L, Steenbergen RD, Rosl F.Alternative splicing of human papillomavirus type-16 E6/E6* early mRNA is coupled to EGF signaling via Erk1/2 activation. Proc Natl Acad Sci USA2010;107: 7006–7011.
- Kozak M.Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol Cell Biol1987;7: 3438–3445.
- Suprynowicz FA, Krawczyk E, Hebert JD et al.The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J Virol2010;84: 10619–10629.
- Pim D, Collins M, Banks L.Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene1992;7: 27–32.
- Rodriguez MI, Finbow ME, Alonso A.Binding of human papillomavirus 16 E5 to the 16 kDa subunit c (proteolipid) of the vacuolar H+-ATPase can be dissociated from the E5-mediated epidermal growth factor receptor overactivation. Oncogene2000;19: 3727–3732.
- Straight SW, Hinkle PM, Jewers RJ, McCance DJ.The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol1993;67: 4521–4532.
- Bodaghi S, Jia R, Zheng ZM.Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology2009;386: 32–43.
- Feng H, Shuda M, Chang Y, Moore PS.Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science2008;319: 1096–1100.
- Shuda M, Feng H, Kwun HJ et al.T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA2008;105: 16272–16277.
- Chang Y, Moore PS.Merkel cell carcinoma: a virus-induced human cancer. Annu Rev Pathol2012;7: 123–144.
- Gazdar AF, Butel JS, Carbone M.SV40 and human tumours: myth, association or causality? Nat Rev Cancer2002;2: 957–964.
- Kwun HJ, Guastafierro A, Shuda M et al.The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J Virol2009;83: 12118–12128.
- Houben R, Adam C, Baeurle A et al.An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int J Cancer2012;130: 847–856.
- Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS.Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest2011;121: 3623–3634.
- Liu X, Hein J, Richardson SC et al.Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human Vam6p from the cytoplasm to the nucleus. J Biol Chem2011;286: 17079–17090.
- Yu Y, Kudchodkar SB, Alwine JC.Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J Virol2005;79: 6882–6889.
- Kwun HJ, Shuda M, Feng H, Camacho CJ, Moore PS, Chang Y.Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe2013;14: 125–135.
- Bhatia K, Goedert JJ, Modali R, Preiss L, Ayers LW.Immunological detection of viral large T antigen identifies a subset of Merkel cell carcinoma tumors with higher viral abundance and better clinical outcome. Int J Cancer2010;127: 1493–1496.
- Houben R, Shuda M, Weinkam R et al.Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol2010;84: 7064–7072.
- Henle W, Henle G.Epidemiologic aspects of Epstein–Barr virus (EBV)-associated diseases. Ann NY Acad Sci1980;354: 326–331.
- Young LS, Rickinson AB.Epstein–Barr virus: 40 years on. Nat Rev Cancer2004;4: 757–768.
- HENLE G, Henle W, Clifford P et al.Antibodies to Epstein–Barr virus in Burkitt's lymphoma and control groups. J Natl Cancer Inst1969;43: 1147–1157.
- Arvey A, Tempera I, Tsai K et al.An atlas of the Epstein–Barr virus transcriptome and epigenome reveals host–virus regulatory interactions. Cell Host Microbe2012;12: 233–245.
- Jones JF, Shurin S, Abramowsky C et al.T-cell lymphomas containing Epstein–Barr viral DNA in patients with chronic Epstein–Barr virus infections. N Engl J Med1988;318: 733–741.
- Jaffe ES, Krenacs L, Raffeld M.Classification of cytotoxic T-cell and natural killer cell lymphomas. Semin Hematol2003;40: 175–184.
- Su IJ, Chen JY.The role of Epstein–Barr virus in lymphoid malignancies. Crit Rev Oncol Hematol1997;26: 25–41.
- Wang D, Liebowitz D, Kieff E.An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell1985;43: 831–840.
- Dirmeier U, Neuhierl B, Kilger E, Reisbach G, Sandberg ML, Hammerschmidt W.Latent membrane protein 1 is critical for efficient growth transformation of human B cells by Epstein–Barr virus. Cancer Res2003;63: 2982–2989.
- Lee DY, Sugden B.The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood2008;111: 2280–2289.
- Humme S, Reisbach G, Feederle R et al.The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci USA2003;100: 10989–10994.
- Kang MS, Soni V, Bronson R, Kieff E.Epstein–Barr virus nuclear antigen 1 does not cause lymphoma in C57BL/6J mice. J Virol2008;82: 4180–4183.
- Decaussin G, Sbih-Lammali F, Turenne-Tessier M, Bouguermouh A, Ooka T.Expression of BARF1 gene encoded by Epstein–Barr virus in nasopharyngeal carcinoma biopsies. Cancer Res2000;60: 5584–5588.
- Wei MX, Ooka T.A transforming function of the BARF1 gene encoded by Epstein–Barr virus. EMBO J1989;8: 2897–2903.
- Seto E, Ooka T, Middeldorp J, Takada K.Reconstitution of nasopharyngeal carcinoma-type EBV infection induces tumorigenicity. Cancer Res2008;68: 1030–1036.
- Sheng W, Decaussin G, Ligout A, Takada K, Ooka T.Malignant transformation of Epstein–Barr virus-negative Akata cells by introduction of the BARF1 gene carried by Epstein–Barr virus. J Virol2003;77: 3859–3865.
- Danve C, Decaussin G, Busson P, Ooka T.Growth transformation of primary epithelial cells with a NPC-derived Epstein–Barr virus strain. Virology2001;288: 223–235.
- Fennewald S, van Santen V, Kieff E.Nucleotide sequence of an mRNA transcribed in latent growth-transforming virus infection indicates that it may encode a membrane protein. J Virol1984;51: 411–419.
- Renzette N, Somasundaran M, Brewster F et al.Epstein–Barr virus latent membrane protein 1 genetic variability in peripheral blood B cells and oropharyngeal fluids. J Virol2014;88: 3744–3755.
- Pandya J, Walling DM.Oncogenic activity of Epstein–Barr virus latent membrane protein 1 (LMP-1) is down-regulated by lytic LMP-1. J Virol2006;80: 8038–8046.
- Kilger E, Kieser A, Baumann M, Hammerschmidt W.Epstein–Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J1998;17: 1700–1709.
- Pratt ZL, Zhang J, Sugden B.The latent membrane protein 1 (LMP1) oncogene of Epstein–Barr virus can simultaneously induce and inhibit apoptosis in B cells. J Virol2012;86: 4380–4393.
- Rowe M, Peng-Pilon M, Huen DS et al.Upregulation of bcl-2 by the Epstein–Barr virus latent membrane protein LMP1: a B-cell-specific response that is delayed relative to NF-kappa B activation and to induction of cell surface markers. J Virol1994;68: 5602–5612.
- Wang S, Rowe M, Lundgren E.Expression of the Epstein Barr virus transforming protein LMP1 causes a rapid and transient stimulation of the Bcl-2 homologue Mcl-1 levels in B-cell lines. Cancer Res1996;56: 4610–4613.
- D'Souza BN, Edelstein LC, Pegman PM et al.Nuclear factor kappa B-dependent activation of the antiapoptotic bfl-1 gene by the Epstein–Barr virus latent membrane protein 1 and activated CD40 receptor. J Virol2004;78: 1800–1816.
- Fries KL, Miller WE, Raab-Traub N.Epstein–Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J Virol1996;70: 8653–8659.
- Bentz GL, Bheda-Malge A, Wang L, Shackelford J, Damania B, Pagano JS.KSHV LANA and EBV LMP1 induce the expression of UCH-L1 following viral transformation. Virology2014;448: 293–302.
- Grimm T, Schneider S, Naschberger E et al.EBV latent membrane protein-1 protects B cells from apoptosis by inhibition of BAX. Blood2005;105: 3263–3269.
- Tsai SC, Lin SJ, Lin CJ et al.Autocrine CCL3 and CCL4 induced by the oncoprotein LMP1 promote Epstein–Barr virus-triggered B cell proliferation. J Virol2013;87: 9041–9052.
- Chen CC, Liu HP, Chao M et al.NF-kappaB-mediated transcriptional upregulation of TNFAIP2 by the Epstein–Barr virus oncoprotein, LMP1, promotes cell motility in nasopharyngeal carcinoma. Oncogene2014;33: 3648–3659.
- Anastasiadou E, Boccellato F, Vincenti S et al.Epstein–Barr virus encoded LMP1 downregulates TCL1 oncogene through miR-29b. Oncogene2010;29: 1316–1328.
- Yu H, Lu J, Zuo L et al.Epstein–Barr virus downregulates microRNA 203 through the oncoprotein latent membrane protein 1: a contribution to increased tumor incidence in epithelial cells. J Virol2012;86: 3088–3099.
- Komabayashi Y, Kishibe K, Nagato T, Ueda S, Takahara M, Harabuchi Y.Downregulation of miR-15a due to LMP1 promotes cell proliferation and predicts poor prognosis in nasal NK/T-cell lymphoma. Am J Hematol2014;89: 25–33.
- Baer R, Bankier AT, Biggin MD et al.DNA sequence and expression of the B95-8 Epstein–Barr virus genome. Nature1984;310: 207–211.
- Hudson GS, Farrell PJ, Barrell BG.Two related but differentially expressed potential membrane proteins encoded by the EcoRI Dhet region of Epstein–Barr virus B95-8. J Virol1985;53: 528–535.
- Vazirabadi G, Geiger TR, Coffin WF III, Martin JM.Epstein–Barr virus latent membrane protein-1 (LMP-1) and lytic LMP-1 localization in plasma membrane-derived extracellular vesicles and intracellular virions. J Gen Virol2003;84: 1997–2008.
- Erickson KD, Berger C, Coffin WF, III, Schiff E, Walling DM, Martin JM.Unexpected absence of the Epstein–Barr virus (EBV) lyLMP-1 open reading frame in tumor virus isolates: lack of correlation between Met129 status and EBV strain identity. J Virol2003;77: 4415–4422.
- Pandya J, Walling DM.Epstein–Barr virus latent membrane protein 1 (LMP-1) half-life in epithelial cells is down-regulated by lytic LMP-1. J Virol2004;78: 8404–8410.
- Concha M, Wang X, Cao S et al.Identification of new viral genes and transcript isoforms during Epstein–Barr virus reactivation using RNA-Seq. J Virol2012;86: 1458–1467.
- Wang D, Liebowitz D, Wang F et al.Epstein–Barr virus latent infection membrane protein alters the human B-lymphocyte phenotype: deletion of the amino terminus abolishes activity. J Virol1988;62: 4173–4184.
- Erickson KD, Martin JM.The late lytic LMP-1 protein of Epstein–Barr virus can negatively regulate LMP-1 signaling. J Virol2000;74: 1057–1060.
- Verma D, Bais S, Gaillard M, Swaminathan S.Epstein–Barr virus SM protein utilizes cellular splicing factor SRp20 to mediate alternative splicing. J Virol2010;84: 11781–11789.
- Juillard F, Bazot Q, Mure F et al.Epstein–Barr virus protein EB2 stimulates cytoplasmic mRNA accumulation by counteracting the deleterious effects of SRp20 on viral mRNAs. Nucleic Acids Res2012;40: 6834–6849.
- Jia R, Li C, McCoy JP, Deng CX, Zheng ZM.SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci2010;6: 806–826.
- Cleghorn FR, Manns A, Falk R et al.Effect of human T-lymphotropic virus type I infection on non-Hodgkin's lymphoma incidence. J Natl Cancer Inst1995;87: 1009–1014.
- Yamaguchi K, Takatsuki K.Adult T cell leukaemia–lymphoma. Baillieres Clin Haematol1993;6: 899–915.
- Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H.Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood1977;50: 481–492.
- Takatsuki K.Discovery of adult T-cell leukemia. Retrovirology2005;2: 16.
- Yoshida M, Miyoshi I, Hinuma Y.Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci USA1982;79: 2031–2035.
- Seiki M, Hattori S, Hirayama Y, Yoshida M.Human adult T-cell leukemia virus: complete nucleotide sequence of the provirus genome integrated in leukemia cell DNA. Proc Natl Acad Sci USA1983;80: 3618–3622.
- Kalyanaraman VS, Narayanan R, Feorino P et al.Isolation and characterization of a human T cell leukemia virus type II from a hemophilia—a patient with pancytopenia. EMBO J1985;4: 1455–1460.
- Calattini S, Chevalier SA, Duprez R et al.Discovery of a new human T-cell lymphotropic virus (HTLV-3) in Central Africa. Retrovirology2005;2: 30.
- Wolfe ND, Heneine W, Carr JK et al.Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc Natl Acad Sci USA2005;102: 7994–7999.
- Grassmann R, Aboud M, Jeang KT.Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene2005;24: 5976–5985.
- Hasegawa H, Sawa H, Lewis MJ et al.Thymus-derived leukemia–lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat Med2006;12: 466–472.
- Cann AJ, Rosenblatt JD, Wachsman W, Shah NP, Chen IS.Identification of the gene responsible for human T-cell leukaemia virus transcriptional regulation. Nature1985;318: 571–574.
- Felber BK, Paskalis H, Kleinman-Ewing C, Wong-Staal F, Pavlakis GN.The pX protein of HTLV-I is a transcriptional activator of its long terminal repeats. Science1985;229: 675–679.
- Sodroski J, Rosen C, Wong-Staal F et al.Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science1985;227: 171–173.
- Nicot C, Harrod RL, Ciminale V, Franchini G.Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene2005;24: 6026–6034.
- Goren I, Semmes OJ, Jeang KT, Moelling K.The amino terminus of Tax is required for interaction with the cyclic AMP response element binding protein. J Virol1995;69: 5806–5811.
- Adya N, Giam CZ.Distinct regions in human T-cell lymphotropic virus type I tax mediate interactions with activator protein CREB and basal transcription factors. J Virol1995;69: 1834–1841.
- Baranger AM, Palmer CR, Hamm MK et al.Mechanism of DNA-binding enhancement by the human T-cell leukaemia virus transactivator Tax. Nature1995;376: 606–608.
- Kwok RP, Laurance ME, Lundblad JR et al.Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature1996;380: 642–646.
- Giebler HA, Loring JE, van Orden K et al.Anchoring of CREB binding protein to the human T-cell leukemia virus type 1 promoter: a molecular mechanism of Tax transactivation. Mol Cell Biol1997;17: 5156–5164.
- Bex F, Yin MJ, Burny A, Gaynor RB.Differential transcriptional activation by human T-cell leukemia virus type 1 Tax mutants is mediated by distinct interactions with CREB binding protein and p300. Mol Cell Biol1998;18: 2392–2405.
- Jiang H, Lu H, Schiltz RL et al.PCAF interacts with tax and stimulates tax transactivation in a histone acetyltransferase-independent manner. Mol Cell Biol1999;19: 8136–8145.
- Harrod R, Kuo YL, Tang Y et al.p300 and p300/cAMP-responsive element-binding protein associated factor interact with human T-cell lymphotropic virus type-1 Tax in a multi-histone acetyltransferase/activator-enhancer complex. J Biol Chem2000;275: 11852–11857.
- Fujii M, Tsuchiya H, Chuhjo T, Akizawa T, Seiki M.Interaction of HTLV-1 Tax1 with p67SRF causes the aberrant induction of cellular immediate early genes through CArG boxes. Genes Dev1992;6: 2066–2076.
- Fujii M, Tsuchiya H, Chuhjo T, Minamino T, Miyamoto K, Seiki M.Serum response factor has functional roles both in indirect binding to the CArG box and in the transcriptional activation function of human T-cell leukemia virus type I Tax. J Virol1994;68: 7275–7283.
- Fujii M, Chuhjo T, Minamino T, Masaaki N, Miyamoto K, Seiki M.Identification of the Tax interaction region of serum response factor that mediates the aberrant induction of immediate early genes through CArG boxes by HTLV-I Tax. Oncogene1995;11: 7–14.
- Ballard DW, Bohnlein E, Lowenthal JW, Wano Y, Franza BR, Greene WC.HTLV-I tax induces cellular proteins that activate the kappa B element in the IL-2 receptor alpha gene. Science1988;241: 1652–1655.
- Inoue J, Seiki M, Taniguchi T, Tsuru S, Yoshida M.Induction of interleukin 2 receptor gene expression by p40x encoded by human T-cell leukemia virus type 1. EMBO J1986;5: 2883–2888.
- Maruyama M, Shibuya H, Harada H et al.Evidence for aberrant activation of the interleukin-2 autocrine loop by HTLV-1-encoded p40x and T3/Ti complex triggering. Cell1987;48: 343–350.
- Kronke M, Leonard WJ, Depper JM, Greene WC.Deregulation of interleukin-2 receptor gene expression in HTLV-I-induced adult T-cell leukemia. Science1985;228: 1215–1217.
- Azimi N, Brown K, Bamford RN, Tagaya Y, Siebenlist U, Waldmann TA.Human T cell lymphotropic virus type I Tax protein trans-activates interleukin 15 gene transcription through an NF-kappaB site. Proc Natl Acad Sci USA1998;95: 2452–2457.
- Mariner JM, Lantz V, Waldmann TA, Azimi N.Human T cell lymphotropic virus type I Tax activates IL-15R alpha gene expression through an NF-kappa B site. J Immunol2001;166: 2602–2609.
- Pise-Masison CA, Mahieux R, Radonovich M, Jiang H, Brady JN.Human T-lymphotropic virus type I Tax protein utilizes distinct pathways for p53 inhibition that are cell type-dependent. J Biol Chem2001;276: 200–205.
- Kaida A, Ariumi Y, Ueda Y et al.Functional impairment of p73 and p51, the p53-related proteins, by the human T-cell leukemia virus type 1 Tax oncoprotein. Oncogene2000;19: 827–830.
- Lemasson I, Nyborg JK.Human T-cell leukemia virus type I tax repression of p73beta is mediated through competition for the C/H1 domain of CBP. J Biol Chem2001;276: 15720–15727.
- Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M.HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene2013;32: 4222–4230.
- Mocquet V, Neusiedler J, Rende F et al.The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J Virol2012;86: 7530–7543.
- Kinoshita T, Shimoyama M, Tobinai K et al.Detection of mRNA for the tax1/rex1 gene of human T-cell leukemia virus type I in fresh peripheral blood mononuclear cells of adult T-cell leukemia patients and viral carriers by using the polymerase chain reaction. Proc Natl Acad Sci USA1989;86: 5620–5624.
- Furukawa Y, Osame M, Kubota R, Tara M, Yoshida M.Human T-cell leukemia virus type-1 (HTLV-1) Tax is expressed at the same level in infected cells of HTLV-1-associated myelopathy or tropical spastic paraparesis patients as in asymptomatic carriers but at a lower level in adult T-cell leukemia cells. Blood1995;85: 1865–1870.
- Ohshima K, Kikuchi M, Masuda Y et al.Defective provirus form of human T-cell leukemia virus type I in adult T-cell leukemia/lymphoma: clinicopathological features. Cancer Res1991;51: 4639–4642.
- Murata K, Hayashibara T, Sugahara K et al.A novel alternative splicing isoform of human T-cell leukemia virus type 1 bZIP factor (HBZ-SI) targets distinct subnuclear localization. J Virol2006;80: 2495–2505.
- Taylor JM, Nicot C.HTLV-1 and apoptosis: role in cellular transformation and recent advances in therapeutic approaches. Apoptosis2008;13: 733–747.
- Russo JJ, Bohenzky RA, Chien MC et al.Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci USA1996;93: 14862–14867.
- Wang HW, Trotter MW, Lagos D et al.Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat Genet2004;36: 687–693.
- Flore O, Rafii S, Ely S, O'Leary JJ, Hyjek EM, Cesarman E.Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature1998;394: 588–592.
- Lunardi-Iskandar Y, Gill P, Lam VH et al.Isolation and characterization of an immortal neoplastic cell line (KS Y-1) from AIDS-associated Kaposi's sarcoma. J Natl Cancer Inst1995;87: 974–981.
- Myoung J, Ganem D.Infection of lymphoblastoid cell lines by Kaposi's sarcoma-associated herpesvirus: critical role of cell-associated virus. J Virol2011;85: 9767–9777.
- Sturzl M, Gaus D, Dirks WG, Ganem D, Jochmann R.Kaposi's sarcoma-derived cell line SLK is not of endothelial origin, but is a contaminant from a known renal carcinoma cell line. Int J Cancer2013;132: 1954–1958.
- Jones T, Ye F, Bedolla R et al.Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J Clin Invest2012;122: 1076–1081.
- Renne R, Zhong W, Herndier B et al.Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med1996;2: 342–346.
- Cannon JS, Ciufo D, Hawkins AL et al.A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J Virol2000;74: 10187–10193.
- Rappocciolo G, Hensler HR, Jais M et al.Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J Virol2008;82: 4793–4806.
- Myoung J, Ganem D.Active lytic infection of human primary tonsillar B cells by KSHV and its noncytolytic control by activated CD4+ T cells. J Clin Invest2011;121: 1130–1140.
- Dollery SJ, Santiago-Crespo RJ, Kardava L, Moir S, Berger EA.Efficient infection of a human B cell line with cell-free Kaposi's sarcoma-associated herpesvirus. J Virol2014;88: 1748–1757.
- Vieira J, O'Hearn PM.Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology2004;325: 225–240.
- Wang LX, Kang G, Kumar P et al.Humanized-BLT mouse model of Kaposi's sarcoma-associated herpesvirus infection. Proc Natl Acad Sci USA2014;111: 3146–3151.
- Si H, Robertson ES.Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J Virol2006;80: 697–709.
- Baresova P, Pitha PM, Lubyova B.Distinct roles of Kaposi's sarcoma-associated herpesvirus-encoded viral interferon regulatory factors in inflammatory response and cancer. J Virol2013;87: 9398–9410.
- Shamay M, Liu J, Li R et al.A protein array screen for Kaposi's sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J Virol2012;86: 5179–5191.
- Godden-Kent D, Talbot SJ, Boshoff C et al.The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol1997;71: 4193–4198.
- Sarek G, Jarviluoma A, Ojala PM.KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood2006;107: 725–732.
- Zhi H, Zahoor MA, Shudofsky AM, Giam CZ.KSHV vCyclin counters the senescence/G1 arrest response triggered by NF-kappaB hyperactivation. Oncogene2014 Jan 27.doi:https://doi.org/10.1038/onc.2013.567.E-pub ahead of print.
- Guasparri I, Keller SA, Cesarman E.KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med2004;199: 993–1003.
- Wang S, Wang S, Maeng H et al.K1 protein of human herpesvirus 8 suppresses lymphoma cell Fas-mediated apoptosis. Blood2007;109: 2174–2182.
- Bais C, Van Geelen A, Eroles P et al.Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell2003;3: 131–143.
- Wang Y, Lu X, Zhu L et al.IKK epsilon kinase is crucial for viral G protein-coupled receptor tumorigenesis. Proc Natl Acad Sci USA2013;110: 11139–11144.
- Tomlinson CC, Damania B.The K1 protein of Kaposi's sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol2004;78: 1918–1927.
- Prakash O, Tang ZY, Peng X et al.Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J Natl Cancer Inst2002;94: 926–935.
- Montaner S, Sodhi A, Molinolo A et al.Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell2003;3: 23–36.
- Sin SH, Dittmer DP.Viral latency locus augments B-cell response in vivo to induce chronic marginal zone enlargement, plasma cell hyperplasia, and lymphoma. Blood2013;121: 2952–2963.
- Pearce M, Matsumura S, Wilson AC.Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J Virol2005;79: 14457–14464.
- Bieleski L, Talbot SJ.Kaposi's sarcoma-associated herpesvirus vCyclin open reading frame contains an internal ribosome entry site. J Virol2001;75: 1864–1869.
- Majerciak V, Ni T, Yang W, Meng B, Zhu J, Zheng ZM.A viral genome landscape of RNA polyadenylation from KSHV latent to lytic infection. PLoS Pathog2013;9: e1003749.
- Cai X, Cullen BR.Transcriptional origin of Kaposi's sarcoma-associated herpesvirus microRNAs. J Virol2006;80: 2234–2242.
- Yan H, Zhong G, Xu G et al.Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife2012;1: e00049.
- Nie J, Li J, Sun K et al.HBV/D1: a major HBV subgenotype circulating in Uyghur patients with chronic HBV infection in Xinjiang, China. Arch Virol2012;157: 1541–1549.
- Inui A, Komatsu H, Sogo T, Nagai T, Abe K, Fujisawa T.Hepatitis B virus genotypes in children and adolescents in Japan: before and after immunization for the prevention of mother to infant transmission of hepatitis B virus. J Med Virol2007;79: 670–675.
- Coursaget P, Yvonnet B, Chotard J et al.Age- and sex-related study of hepatitis B virus chronic carrier state in infants from an endemic area (Senegal). J Med Virol1987;22: 1–5.
- Benhenda S, Ducroux A, Riviere L et al.Methyltransferase PRMT1 is a binding partner of HBx and a negative regulator of hepatitis B virus transcription. J Virol2013;87: 4360–4371.
- Lucito R, Schneider RJ.Hepatitis B virus X protein activates transcription factor NF-kappa B without a requirement for protein kinase C. J Virol1992;66: 983–991.
- Lian Z, Liu J, Wu M et al.Hepatitis B x antigen up-regulates vascular endothelial growth factor receptor 3 in hepatocarcinogenesis. Hepatology2007;45: 1390–1399.
- Arzumanyan A, Friedman T, Ng IO, Clayton MM, Lian Z, Feitelson MA.Does the hepatitis B antigen HBx promote the appearance of liver cancer stem cells? Cancer Res2011;71: 3701–3708.
- Lu JW, Yang WY, Lin YM, Jin SL, Yuh CH.Hepatitis B virus X antigen and aflatoxin B1 synergistically cause hepatitis, steatosis and liver hyperplasia in transgenic zebrafish. Acta Histochem2013;115: 728–739.
- Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P.Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature1979;281: 646–650.
- Antonucci TK, Rutter WJ.Hepatitis B virus (HBV) promoters are regulated by the HBV enhancer in a tissue-specific manner. J Virol1989;63: 579–583.
- Zheng Y, Li J, Ou JH.Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J Virol2004;78: 6908–6914.
- Sommer G, Heise T.Posttranscriptional control of HBV gene expression. Front Biosci2008;13: 5533–5547.
- Schwalbe M, Ohlenschlager O, Marchanka A et al.Solution structure of stem-loop alpha of the hepatitis B virus post-transcriptional regulatory element. Nucleic Acids Res2008;36: 1681–1689.
- Huang C, Xie MH, Liu W et al.A structured RNA in hepatitis B virus post-transcriptional regulatory element represses alternative splicing in a sequence-independent and position-dependent manner. FEBS J2011;278: 1533–1546.
- Chowdhury JB, Roy D, Ghosh S.Identification of a unique splicing regulatory cluster in hepatitis B virus pregenomic RNA. FEBS Lett2011;585: 3348–3353.
- Heise T, Sommer G, Reumann K, Meyer I, Will H, Schaal H.The hepatitis B virus PRE contains a splicing regulatory element. Nucleic Acids Res2006;34: 353–363.
- Soussan P, Garreau F, Zylberberg H, Ferray C, Brechot C, Kremsdorf D.In vivo expression of a new hepatitis B virus protein encoded by a spliced RNA. J Clin Invest2000;105: 55–60.
- Bayliss J, Lim L, Thompson AJ et al.Hepatitis B virus splicing is enhanced prior to development of hepatocellular carcinoma. J Hepatol2013;59: 1022–1028.
- Soussan P, Tuveri R, Nalpas B et al.The expression of hepatitis B spliced protein (HBSP) encoded by a spliced hepatitis B virus RNA is associated with viral replication and liver fibrosis. J Hepatol2003;38: 343–348.
- Chen WN, Chen JY, Lin WS, Lin JY, Lin X.Hepatitis B doubly spliced protein, generated by a 2.2 kb doubly spliced hepatitis B virus RNA, is a pleiotropic activator protein mediating its effects via activator protein-1- and CCAAT/enhancer-binding protein-binding sites. J Gen Virol2010;91: 2592–2600.
- Chen WN, Chen JY, Jiao BY et al.Interaction of the hepatitis B spliced protein with cathepsin B promotes hepatoma cell migration and invasion. J Virol2012;86: 13533–13541.
- Mancini-Bourgine M, Bayard F, Soussan P et al.Hepatitis B virus splice-generated protein induces T-cell responses in HLA-transgenic mice and hepatitis B virus-infected patients. J Virol2007;81: 4963–4972.
- Ma ZM, Lin X, Wang YX, Tian XC, Xie YH, Wen YM.A double-spliced defective hepatitis B virus genome derived from hepatocellular carcinoma tissue enhanced replication of full-length virus. J Med Virol2009;81: 230–237.
- Huang HL, Jeng KS, Hu CP, Tsai CH, Lo SJ, Chang C.Identification and characterization of a structural protein of hepatitis B virus: a polymerase and surface fusion protein encoded by a spliced RNA. Virology2000;275: 398–410.
- Park GS, Kim HY, Shin HS, Park S, Shin HJ, Kim K.Modulation of hepatitis B virus replication by expression of polymerase-surface fusion protein through splicing: implications for viral persistence. Virus Res2008;136: 166–174.
- Garcia-Sastre A, Evans MJ.miR-122 is more than a shield for the hepatitis C virus genome. Proc Natl Acad Sci USA2013;110: 1571–1572.
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P.Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science2005;309: 1577–1581.
- Steinmann E, Pietschmann T.Cell culture systems for hepatitis C virus. Curr Top Microbiol Immunol2013;369: 17–48.
- Wakita T, Pietschmann T, Kato T et al.Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med2005;11: 791–796.
- Zhong J, Gastaminza P, Cheng G et al.Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA2005;102: 9294–9299.
- Levrero M.Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene2006;25: 3834–3847.
- Banerjee A, Ray RB, Ray R.Oncogenic potential of hepatitis C virus proteins. Viruses2010;2: 2108–2133.
- Akkari L, Gregoire D, Floc'h N et al.Hepatitis C viral protein NS5A induces EMT and participates in oncogenic transformation of primary hepatocyte precursors. J Hepatol2012;57: 1021–1028.
- Kowdley KV, Gordon SC, Reddy KR et al.Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med2014;370: 1879–1888.
- Gane EJ, Stedman CA, Hyland RH et al.Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A Inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology2014;146: 736–743.
- Lawitz E, Poordad FF, Pang PS et al.Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet2014;383: 515–523.