
Abstract
We report the discovery and confirmation of 23 novel mutations with previously undocumented role in isoniazid (INH) drug resistance, in catalase-peroxidase (katG) gene of Mycobacterium tuberculosis (Mtb) isolates. With these mutations, a synonymous mutation in fabG1g609a, and two canonical mutations, we were able to explain 98% of the phenotypic resistance observed in 366 clinical Mtb isolates collected from four high tuberculosis (TB)-burden countries: India, Moldova, Philippines, and South Africa. We conducted overlapping targeted and whole-genome sequencing for variant discovery in all clinical isolates with a variety of INH-resistant phenotypes. Our analysis showed that just two canonical mutations (katG 315AGC-ACC and inhA promoter-15C-T) identified 89.5% of resistance phenotypes in our collection. Inclusion of the 23 novel mutations reported here, and the previously documented point mutation in fabG1, increased the sensitivity of these mutations as markers of INH resistance to 98%. Only six (2%) of the 332 resistant isolates in our collection did not harbor one or more of these mutations. The third most prevalent substitution, at inhA promoter position -8, present in 39 resistant isolates, was of no diagnostic significance since it always co-occurred with katG 315. 79% of our isolates harboring novel mutations belong to genetic group 1 indicating a higher tendency for this group to go down an uncommon evolutionary path and evade molecular diagnostics. The results of this study contribute to our understanding of the mechanisms of INH resistance in Mtb isolates that lack the canonical mutations and could improve the sensitivity of next generation molecular diagnostics.
Introduction
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB) and was responsible for the death of 1.5 million people in 2013.Citation1 Multi-drug resistant TB (MDR-TB) strains are those that are resistant to at least rifampicin (RIF) and isoniazid (INH)—the two most effective first line drugs used to treat TB.Citation1 Globally, 3.5% of new and 20.5% of previously treated cases (or a total of 300 000 cases) were estimated to be MDR-TB in 2013.Citation1 Increasing resistance levels and rates of treatment failures in certain parts of the world are of major concern.Citation1 Moreover, the availability of reliable and timely methods for the detection of drug-resistant TB strains has been inadequate as growth-based methods such as phenotypic drug susceptibility testing (DST) take weeks to months to complete and require laboratory infrastructure that is not widely available in countries with a high burden of TB.Citation1,Citation2 The World Health Organization (WHO) estimates that only about 45% of all MDR-TB cases were diagnosed and notified in 2013.Citation1 In part due to new rapid diagnostic methods, this rate has been continuously increasing from nearly 17% in 2009.Citation1
Molecular diagnostics such as GeneXpert and Hain GenoType MTBDRplus line probe assay (LPA),Citation3 which have vastly simplified and increased the speed of diagnosing drug-resistant TB, appear to suffer from variable sensitivity and specificity depending on the geographic region of the world in which they are employed.Citation4 More importantly, GeneXpert only detects RIF resistance, while the line-probe does not have the full spectrum of probes to detect all INH resistance phenotypes (INHR). In some regions, RIF resistance is used as a marker for MDR-TB. This method, however, misses the INH-monoresistant cases that could evolve into MDR-TB.Citation5 Mono-INHR is thought to be on the rise in certain geographic regions—possibly as a consequence of failures to detect monoresistant strains with molecular diagnostics.Citation6 Additionally, there is evidence to suggest that the frequency of the S315T mutation, most commonly documented to confer INHR, is far lower in mono-INHR isolates (25%) than in MDR-TB isolates (79%), making a broader search for novel INHR-conferring mutations to include in next generation molecular diagnostics that much more critical.Citation5,Citation6
Clinical significance of INH resistance is most commonly due to missense mutations within the katG gene. Frequently, mutated S315T katG produces a functional catalase-peroxidase (katG) with enzymatic activity while highly deficient in its ability to form an INH-nicotinamide adenine dinucleotide adduct, reducing the INH toxicity and having little fitness cost to the bacterium.Citation7,Citation8,Citation9 Alternatively, low-level resistance can emerge due to point mutations in the regulatory region of the inhA operon.Citation10 For example, -15C-T mutation within the inhA promoter region, has been observed to increase inhA mRNA level to 20 times that of the wild type, resulting in inhA overexpression and an eight-fold increase in resistance to INH.Citation11 While two polymorphisms, katG (S315T) and inhA (-15C-T), explain the majority of INH resistance in clinical isolates,Citation10,Citation12 other mutations carry the explanation for a subset of INHR cases that lack the two most common canonical mutations.Citation13,Citation14
Alternative mechanisms of INHR are not confined to katG and inhA. For instance, a synonymous mutation fabG1 L203L (CTG-CTA) (or fabG1g609a) was first associated with INH resistance by Ramaswamy et al. in 2003.Citation15 In 2014, Ando et al. described the role of this mutation as one that converts a segment of fabG1 into an alternative promoter for inhA and hence increases its expression level, suggesting these mutations may be important for next generation molecular diagnostics.Citation16
In this study, we considered 366 clinical isolates collected in a joint effort through the Global Consortium for Drug-resistant TB Diagnostics (GCDD)Citation17 from four high burden TB countries: India, Moldova, Philippines, and South Africa. We have identified 23 novel katG mutations associated with INH resistance and used mutagenesis to confirm the causal role of those that appeared in strains without a canonical mutation. Our results also indicate that genetic group 1 strains are more likely to traverse down an uncommon evolutionary path and thereby harbor uncommon mutations and hence more likely to evade molecular diagnostics. We believe the results of this research leads to a greater understanding of the full spectrum of mutations that can cause INH resistance, especially in those isolates that lack the canonical mutations, and therefore, to a more sensitive genotypic diagnostic of INH resistance.
Materials and methods
Sample set
Two sets of clinical isolates were used in this study. The first is referred to as the “archive set”. It contains 346 (316 INHR and 30 INHS) clinical isolates collected and sequenced as part of GCDD from India, Moldova, Philippines, and South Africa. Each isolate was collected from a different patient and therefore the set represents 346 independent isolates from the four countries. A second set, referred to as the “supplemental set” contained 20 additional clinical isolates (16 INHR and 4 INHS). These isolates were specifically selected because they harbored no canonical mutations in katG or the inhA promoter. In this article, we define canonical mutations as those occurring in codon 315 of katG or in positions -17, -15, or -8 of inhA promoter region. In total, this study considered 366 (332 INHR and 34 INHS) clinical isolates.
Archive sample collection strategy
The goal of GCDD was to obtain a large collection of M/extensively drug-resistant (XDR)-TB isolates that maximized diversity of M/XDR-TB phenotypes (i.e. DST profiles). Based on the global prevalence of M/XDR-TB and availability of large repositories of M/XDR-TB isolates, we focused our collection on India, Moldova, Philippines, and South Africa. For this study, we sequenced 316 INHR and 30 INHS of these isolates. INH resistance was determined using DST as described below. Details of sample selection and phenotypic characterization of these isolates can be found in Rodwell et al. (2014).Citation12
Supplemental sample collection strategy
The goal of the collection of the supplemental isolates was to identify and study the mechanism of resistance in INHR clinical isolates that lacked any canonical mutations in katG and inhA promoter. INH resistance was determined using DST as described below. The minimum inhibitory concentration (MIC) of each isolate was also estimated as described below. In this set, 16 INHR and 4 INHS clinical isolates without any canonical mutations in katG or inhA were included (all from South Africa).
Sample representativeness
Samples from the archive set were drawn from M/XDR-TB repositories at (i) Hinduja National Hospital (PDHNH) in Mumbai, India representing Mumbai strains; (ii) Phthisiopneumology Institute (PPI) in Chisinau, the central unit of the National TB Control in Moldova, representing national TB strains in Moldova; (iii) Philippine Tropical Disease Foundation (TDF), representing Manila strains; and (iv) The National Health Laboratory Service of South Africa (NHLS), representing a national archive of South African TB strains.
Samples from the supplemental set were drawn from the NHLS similar to the archive set with a selection bias for isolates that had no canonical mutations in katG or inhA promoter region.
Phenotyping
Drug susceptibility testing of archive samples
Upon receiving isolates at University of California, San Diego (UCSD), each isolate was subjected to DST for INH on the Mycobacterial Growth Indicator Tube (MGIT) 960 platform, analyzed by EpiCenter software (BD Diagnostic Systems, Franklin Lakes, NJ, USA), using standard manufacturer protocol and WHO critical concentration (CC) of 0.1 mg/L for INH.Citation18 Using MGIT 960, the DST of the isolates was also determined for six additional first- and second-line drugs (Supplementary Table S1).
Determination of MIC of supplement samples
MICs were determined using MGIT 960 system, according to the agar proportion method, looking for 99% inhibition of growth. Cultures were exposed to INH concentrations of 0.05, 0.1, 0.5, 3, and 10 mg/L. Therefore, an MIC level of 10, indicates a strain with an MIC level of 10 mg/L or greater. Values reported here as “≤10” indicate that there was not complete inhibition of growth (99%) at 10 mg/L of INH, but that the strain was struggling to grow at that concentration despite reaching the required growth units. Isolates reaching 100 growth units within a week of the control were marked as resistant at the concentration. Isolates not reaching 100 growth units were labeled as sensitive at that concentration.
Lineage determination
Lineage typing was performed using spoligotyping and MIRU as described in Garfein et al. (2015).Citation19 In summary, using standardized methods,Citation20,Citation21,Citation22 spoligotyping and 12 mycobacterial interspersed repetitive unit (MIRU-12) assay was determined. TB Insight’s TB-Lineage,Citation23 and online lineage prediction tool was used for the analysis of the resulting Spoligotyping and MIRU-12 results and for lineage prediction using a knowledge-based Bayesian network.Citation24
Sequencing
Whole-genome sequencing (WGS) was used as the primary sequencing technology for this project. The katG gene (2223 bp) and inhA promoter region (289 bp) were first examined for presence or absence of canonical mutations (katG 315, inhA -8, -15 and -17). Since the absence (or presence) of canonical mutations in isolates that harbor novel mutations were critical for this study, we have confirmed the WGS discovery (or lack thereof) of these mutations using the previously published Sanger sequencingCitation12 and PyrosequencingCitation25 results from the same isolates. The WGS data (which cover the entire genes rather than small regions around canonical mutations) are being published here in this article for the first time.
Whole-genome sequencing
Pacific Biosciences single molecule, real-time sequencer (PacBio RS) was used for WGS. Since the Mtb genome has a high GC content, this technology was chosen because it does not use polymerase chain reaction (PCR) amplification and therefor does not suffer from GC bias.
Sample preparation and DNA extraction. Clinical isolates were cultured on Lowenstein Jensen media, killed through exposure to ethanol and heat, lysed, and then DNA extracted.Citation26 The supplemental sample set were grown on 7H10 agar, heat killed and DNA was isolated using the QIAGEN Genomic tip 100/g, cat. nr. 10243.Citation27
Library preparation. DNA libraries for PacBio (Pacific Biosciences, Melon Park, CA, USA) were prepared using PacBio’s DNA Template Prep Kit with no follow-up PCR amplification. Briefly, sheared DNA was end repaired, and hairpin adapters were ligated using T4 DNA ligase. Incompletely formed SMRTbell templates were degraded with a combination of exonuclease III and exonuclease VII. The resulting DNA templates were purified using SPRI magnetic beads (AMPure, Agencourt Bioscience, Beverly, MA, USA) and annealed to a two-fold molar excess of a sequencing primer that specifically bound to the single-stranded loop region of the hairpin adapters. The final sample resulted in library insert sizes of 2 kb in length. SMRTbell templates were subjected to standard SMRT sequencing using an engineered phi29 DNA polymerase on the PacBio RS system according to manufacturer’s protocol.
Post-sequencing analysis. Raw H5 files produced by the RS were processed by an in-house WGS pipeline developed for the Pacific Biosciences RS system. The Pac-DAP (Pacific Biosciences Data Analysis Protocol) pipeline was developed by the Bioinformatics and Medical Informatics Research Center (BMIRC) of San Diego State University (SDSU). It employs a combination of SMRT Portal (version 1.3.3) (PacBio sequencer’s native software), GATK (version 2.2.2),Citation28,Citation29,Citation30 SAMtools (version 0.1.18),Citation31 BamTools (version 2.2.3),Citation32 VarScan (Version 2.3.6),Citation33,Citation34 and in-house developed code for variant calling and housekeeping. In brief, the raw H5 data files of RS were imported into SMRT Portal. Reads from the H5 files were aligned to M. tuberculosis H37Rv genome from NCBI by BLASR, SMRT Portal’s Basic Local Alignment. Quality scores were recalibrated and converted from Pacific Biosciences’ scoring metric into Phred quality scores by using GATK. SAMtools converted recalibrated BAM files into pileup files for VarScan’s variant caller and consensus generator. For the purpose of this study, only single nucleotide polymorphisms (SNPs) were considered and insertions and deletions were ignored as no significant number of insertions or deletions have been previously associated with INH resistance.
Sanger sequencing
Applied Biosystems 3730 XL Analyzer with BigDye Terminator v3.1 Cycle Sequencing Kit was used to sequence 12.3% of the katG gene (274 bases) around codon 315.Citation12 The complete inhA promoter locus was sequenced, roughly 289 bases.Citation12 Forward primers were used for sense strand sequencing and chromatograms were scored by the ABI base caller with the sensitivity set at Q20 (Supplementary Table S2). Any bases scored as “N” were visually read and a base letter was assigned. If the chromatogram was unclear, isolates were excluded. The relevant PCR products for sequencing were generated using the primer pairs as indicated in Supplementary Table S2.
Pyrosequencing
PyroMark Q96 ID system (Qiagen, Valencia, CA, USA) was used to perform Pyrosequencing of Sanger sequencing gene targets identified in katG and inhA promoter. Variant calling in Pyrosequencing was performed automatically with Identifier Software (Qiagen, Valencia, CA, USA) according to manufacturer specifications and procedures as described by Lin et al. (2014).Citation35 Two narrow ranges around katG 315 (codons 312 through 316) and inhA -15 (-4 through -20) positions were sequenced as described in Lin et al. (2014).Citation35 Pyrosequencing was performed at the California Department of Public Health, Microbial Diseases Laboratory.
SNPs discordance resolution
Two regions, 13 bases in katG (codons 312-316) (2155164-2155176) and 17 bases in inhA promoter (-4 to -20), were sequenced by all three technologies.Citation35 Base calling in these regions required the additional condition of at least two of the three sequencing methods having to agree on the call. In this way, variant calling for canonical mutations (katG 315, inhA -8, -15 and -17) required the concordance of at least two of the three platforms.
Functional confirmation of the role of novel mutations in INH resistance
In this study, we have used Mycobacterium smegmatis (due to its faster growth rate) to confirm the role of standalone novel katG mutations in INH resistance. M. smegmatis has been previously used as a surrogate for Mtb in a variety of functional confirmation studies.Citation36,Citation37,Citation38,Citation39,Citation40,Citation41,Citation42,Citation43,Citation44 Xu et al. (2011) used M. smegmatis to confirm the role of mutations in INH and ethionamide resistance.Citation38 Parish et al. (2007) demonstrated that M. smegmatis can be used to assess the functional complementation of fabG1.Citation41 The same study determined that while M. smegmatis is a good surrogate for Mtb, Escherichia coli is not.Citation41
M. smegmatis katG deletion mutant construction
The katG gene from M. smegmatis strain mc2155, encoded by MSMEG_6384 was deleted from its genome by using allelic exchange as previously described.Citation45 Fragments F1 (flanking the 5′ end of katG) and F2 (flanking the 3′ end of katG) were generated using PCR, using primer pair KGF1 (5′-GCC AAG CTC GAC CAG TTG C-3′) and KGR1 (5′-GGT ACT CGG TGC GGA ACT GC-3′), and pair KGF2 (5′-CAA TTC ACC ACT CCC GAA AGC ACA CAA CCA CCT GGA C-3′) and KGR2 (5′-GGT ACT CGG TGC GGA ACT GC-3′) respectively. F1 and F2 were linked together using F1 and F2 PCR products as templates in a PCR with primer pair KGF1 and KGR2, using the region of homology between primers KGR1 and KGF2 to generate a single product. PCR conditions were: initial denaturation at 98°C for 30 s, followed by 35 cycles consisting of denaturation at 98°C for 10 s, hybridization at 60°C for 15 s, and elongation at 72°C for 3 min. Phusion High-fidelity PolymeraseCitation46 was used for all reactions. The resulting product, F1R2, was cloned into the pJET1.2 vector and its identity confirmed with Sanger sequencing. F1R2 was sub-cloned into the allelic exchange vector p2NIL to create plasmid pNKF1R2, where-after the PacI fragment from plasmid pGOAL17 was cloned into the PacI site of pNKF1R2 to create suicide construct pNKF1R2G. This plasmid was electroporated into M. smegmatis mc2155 and mutants selected in a two-step mutagenesis process by single cross-over mutant selection on Luria agar containing hygromycin (50 µg/mL), followed by selection of katG deletion mutants on sucrose-containing agar. Deletion of katG was confirmed with PCR and Southern hybridization, the latter employing labeled fragment F1 as probe and detection with the aid of the ECL Direct Nucleic Acid Labelling and Detection System (Amersham).
Complementation of M. smegmatis katG mutant
Primer pair KGpromF (5′-TGG CGA GGC ACC CTG TCT GAC G-3′) and KGR (5′-GGT GCT GCG GCG GGT TGT GG-3′) and Phusion Polymerase were used to amplify a DNA fragment from M. tuberculosis clinical strain R55. This strain contains the wild-type katG gene with the phylogenetic marker mutation at codon 463. A fragment with this marker was selected specifically because the majority of novel mutations described here contains this marker. PCR was performed using an initial denaturation step at 98°C for 30 s, followed by 35 cycles consisting of denaturation at 98°C for 10 s, hybridization at 60°C for 15 s, and elongation at 72°C for 2.5 min. The product contained the wild-type M. tuberculosis katG gene, encoded by Rv1908c, as well as the promoter directly upstream of the katG gene and was cloned into pJET1.2. Sequence homology was verified with Sanger sequencing before the product was sub-cloned into plasmid mycobacterial integration vector pMV306, to generate integration construct pMV1908c. This construct was used to generate plasmids containing novel katG variants, using the components of the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, California, USA) and the modified site-directed mutagenesis method as described previously.Citation47 The presence of the mutations was confirmed using Sanger sequencing with primers promKGF, RTB59, RTB38, and KGR, enabling sequence verification of the full length gene as well as the promoter. Primers to generate the various mutations are described in Supplementary Table S3. A variant containing the canonical S315T mutation, named pMV-S315T, was also constructed, and used as a resistance control for the phenotypic assay. The various plasmids were transformed into the katG deletion mutant and selected on Luria agar containing hygromycin (50 µg/mL). Plasmids were named pMV-X, with X denoting the number of the mutated codon from M. tuberculosis katG.
Phenotypic characterization of INH resistance in M. smegmatis
M. smegmatis katG wild-type mc2155, katG deletion mutant kGdel, as well as complemented strains were grown overnight in Middlebrook 7H9-OADC (0.2% glycerol, Middlebrook oleic acid-albumin-dextrose-catalase (OADC), and 0.05% Tween 80), to reach an optical density (determined at 600 nm) of 0.8–1. The optical density was standardized to 0.1, where-after serial dilutions of each strain were made in Middlebrook 7H9 broth. Aliquots, 5 µL each, were spotted onto 7H10-OADC agar containing 12 µg/mL of INH. Plates were incubated for two to three days before colonies were visible. The presence of growth was regarded as an indication of resistance, while the absence or very faint growth indicated sensitivity toward INH. The mutant transformed with pMV1908c were used as a control for restoration of sensitivity, while the mutant transformed with pMV-S315T were used as a control for resistance to INH (Supplementary Figures S1–S4).
Results
Drug susceptibility testing
Susceptibility testing of the archive isolates identified 30 INHS and 316 INHR isolates. Seven of the 316 INHR isolates (2.2%) were mono-INH resistant and 301 were MDR-TB, of which 242 were XDR. Details of the resistance phenotypes are available in Supplementary Table S1. As mentioned before, the supplement set contained 16 INHR and four INHS isolates. In total, this study considered 332 INHR and 34 INHS isolates for WGS analysis. With our WGS approach, we were also able to identify polymorphisms in codon 463 allowing categorization of our clinical isolates into distinct lineage and epidemiological genetic groups. Breakdown of the observation of this mutation per country is shown in Supplementary Table S4. 191 of the 332 (or 58%) resistant isolates and 22 of the 34 (or 63%) susceptible isolates harbored a R463L mutation.
Canonical mutations
Two hundred and ninety-eight (89.5%) INHR and two (5.9%) INHS isolates contained one or both of the two (katG 315AGC-ACC and inhA promoter -15C-T) canonical mutations conferring INH resistance. The most frequent mutation found was S315T, which was present in 273 (82%) of resistant strains. Also two susceptible (by DST) strains harbored a canonical mutation (one had katG 315AGC-ACC and harbored inhA promoter -15C-T) (see the “DISCUSSION” section for more information about this discordance). shows the breakdown of the 366 isolates included in this study. The numbers reported in for all canonical mutations have been confirmed by at least two of the three sequencing platforms. Supplementary Figure S5 shows the prevalence of various Mtb lineages among our resistant isolates that harbored a canonical mutation.
Table 1 Isolate stratification based on substitutions in katG and inhA promoter
Other previously characterized katG mutations
Seven resistant (and no susceptible) isolates harbored other previously characterized, non-canonical, resistance-causing katG mutations. Four of the seven also harbored a canonical mutation while three did not and hence the non-canonical mutations observed in the three isolates were the only explanation for resistance in katG or inhA.
Novel mutations
The remaining “unexplained” INHR isolates (32 isolates) were of particular interest because of their lack of canonical and other previously characterized INH resistance causing mutations in katG and inhA promoter region. Within this subset of unexplained INHR isolates, 14 novel katG substitutions were identified in 15 INHR isolates (). It is important to note that a mutation in codon 64 was previously reported by Chan et al. in 2007.Citation48 We believe that this mutation was reported in the reverse direction and therefore not a true representation of codon 64 and the polymorphism we observe within our dataset.
Eleven of the 15 isolates harboring the listed novel katG mutations also harbored R463L (CGG-CTG) (Arg463Leu) polymorphism. The position of eight observed polymorphisms in katG codons 131,142, 162, 269, 306, 387, 394, 541 have been previously reported in INHR isolates,Citation7,Citation49,Citation50,Citation51,Citation52 however the specific polymorphisms we report at the eight loci are novel. These are: P131T(CCG-ACG), D142G(GAC-GGC), A162V(GCG-GTG), G269D(GGT-GAT), T306P(ACC-CCC), D387G(GAT-GGT), T394M(ACG-ATG), A541D(GCC-GAC). Furthermore, our group reported the discovery of T306P by Sanger sequencing in a recent manuscript.Citation12 Here we report that a broader examination of the rest of the katG gene and the inhA promoter region using WGS of the isolate revealed no other resistance associated mutations in inhA or katG genes. We, therefore, describe and confirm (through mutagenesis) here the novel role of this mutation as a new mechanism of resistance.
In addition to the 14 novel katG mutations, nine additional novel katG amino acid substitutions were observed either in combination with katG S315T or with -15C-T inhA promoter mutation (). Two of these substitutions (N596S and Y597H) occurred together in a single isolate that also harbored the inhA -15C-T substitution. Studies have shown that clinical isolates that harbor both the -15C-T inhA promoter and a katG mutation have higher MIC’s than either a single S315T katG or inhA promoter mutation.Citation15 Interestingly, the majority of the novel mutations listed in co-occur with inhA -15 substitution. Only two isolates, harboring D194G and the Y597H–N596S combination, of the nine mutations listed in co-occur with a katG 315 substitution.
In total, 23 unique katG amino acid substitutions were observed, 14 novel standalone katG mutations, an additional 9 novel katG mutations either in combination with S315T or -15C-T inhA promoter mutations. Lastly, seven previously characterized polymorphisms, I335V, G299S, T275A, A264T, E127P, A110V, and A93V were also observed within the collected clinical isolates (Supplementary Table S5).Citation13,Citation14,Citation16,Citation53,Citation54,Citation55,Citation56,Citation57,Citation58,Citation59,Citation60 displays the spatial proximity of the 23 mutations ( and ) to known resistance conferring mutations. A subset of novel standalone katG polymorphisms fell in close proximity to previously described katG substitutions suggesting a similar role in INH resistance ().Citation16,Citation54,Citation55 INH MICs were determined for the 16 resistant isolates from the supplemental set of isolates (Supplementary Table S6). These values are also included (in log scale) in for a visual correlation between the location of the mutations and their associated MIC levels.
Figure 1 Distribution of novel and known katG SNPs.
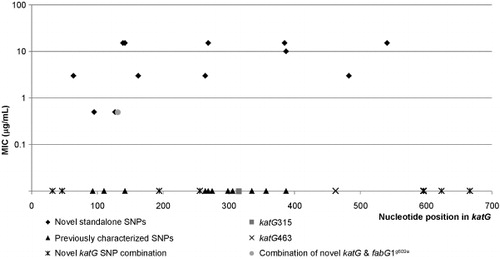
Table 2 Novel katG substitutions in isolates lacking canonical mutations
Mutations in fabG1
The role of the recently described alternative mechanism of INH resistance through fabG1g609a was also investigated among our isolates. Supplementary Table S7 shows the grouping of the isolates that harbored this mutation. Fifteen resistant and no INH susceptible isolates harbored this mutation. Of the 15, 10 harbored a canonical katG 315 mutation but five did not. Of the five, two harbored a novel mutation in katG reported above, namely, P131T and T306P, and one harbored a previously described but rare mutation in katG, namely, G299S. Mutagenesis experiments would be able to elucidate whether the observed resistance in these isolates is solely due to the fabG1 mutation, the katG mutations, or both having an epistatic effect to cause a higher resistance level. Two remaining isolates did not harbor any mutations in katG or inhA promoter other than the fabG1 and the katG463 lineage marker. INH resistance in the remaining two isolates is probably due to fabG1g609a, as no other known INH resistance conferring markers were detected by the three sequencing methods in these previously unexplained resistant cases. Both of these isolates harbor katG463 lineage marker and are from South Africa.
Table 3 Novel katG mutations in isolates that also harbor the canonical mutations
Mutations not conferring resistance
For the completeness of this manuscript, we also report katG mutations observed in INHS isolates, and synonymous katG substitutions. Supplementary Table S8 lists two substitutions in katG gene of INHS isolates. Neither of these substitutions have been previously reported, although a P280H substitution (same position but different substitution as our observed P280S) was reported by Gagneux et al. (2006) as conferring INH resistance.Citation7 Our isolate is mono-RIFR, and therefore we believe our substitution does not confer resistance. Supplementary Table S9 lists the synonymous mutations observed in INHR isolates. Since these synonymous SNPs do not cause any change in the structure of the protein, it is unlikely they confer resistance.
Isolates with no katG, inhA, or fabG1 resistance conferring mutations
As indicates, eight (2%) INHR and 32 (94%) INHS isolates did not harbor any mutations in the three genes studied in this project except for the lineage marker in katG codon 463. The eight resistant isolates are of great interest for discovery of alternative mechanism(s) of resistance (see the “DISCUSSION” section).
Confirmation of causal role in resistance
To verify if the identified novel standalone mutations confer resistance to INH in mycobacteria, we completed a series of mutagenesis experiments in M. smegmatis where we deleted katG, encoded by MSMEG_6384 and showed that the absence of this gene caused it to grow at a higher concentration of INH (Supplementary Figure S1). Complementation of the mutant with the wild-type katG gene from M. tuberculosis restored sensitivity to INH, whereas complementation with a variant encoding S315T did not restore sensitivity to the mutant (Supplementary Figure S2). Similarly, complementation with novel M. tuberculosis katG variants Y95C, P131T, D142G, A162V, T306P, Q439P, F483L, and A541D did not restore sensitivity to INH (Supplementary Figure S3), indicating these mutations confer resistance. A previously described variant, A264T, also did not restore sensitivity to INH, confirming its role in INH resistance.Citation58 In contrast, complementation with a wild-type katG (which contained a polymorphism at 463), or the variants with mutations in codons R385W (Supplementary Figure S3) or D387G (Supplementary Figure S4) restored sensitivity to INH indicating that they do not appear to play a role in INH resistance.
Discussion
From our collection of 332 INHR and 34 INHS clinical isolates, 89.5% of INH resistance could be explained by the two mutations of katG S315T, inhA promoter -15C-T mutations, or a combination of these two mutations. One isolate determined to be susceptible to INH by phenotypic DST harbored the katG S315T mutation and another harbored the inhA promoter -15C-T. The presence of both mutations was confirmed by all three sequencing platforms. We believe that the genotypic–phenotypic discordance may be due to variation in growth around the cut point of DST, although heterogeneity could be another cause of the discordance.
It is interesting to note that the third most prevalent mutation (present in 39 of 332 INHR isolates or 12%) at inhA promoter position -8 appeared to have little diagnostic significance in our isolates as they always co-occurred with katG315. The same is also true for mutations occurring at inhA promoter position -34. Because of the geographic variability of the prevalence of the canonical mutations conferring INH resistance, the sensitivity of molecular diagnostics for detecting INH resistant phenotypes can be variable.Citation4 The discovery of 14 novel, standalone katG mutations could significantly improve the sensitivity of next generation molecular diagnostics that include these mutations, especially in regions where these mutations are more common (e.g. South Africa). In our dataset, using all of the mutations demonstrated to confer resistance, we were able to explain 326 of 332 INHR (98%) of INH resistant phenotypes by a katG, inhA promoter, or a fabG1g609a mutation. This is a notable improvement over the commonly reported sensitivity of molecular diagnostic in the high eighties to low nineties.Citation12,Citation61,Citation62
While all but two canonical mutations, katG S315T and inhA promoter -15C-T, seem not to be useful in diagnostics (since they co-occur with those mutations), we believe that they are important in prognosis of the disease. Previous work has shown that combination of inhA promoter and katG results in higher MIC levels.Citation10,Citation63 We believe the same effect may be taking place for combination of novel mutations reported and canonical mutations in inhA promoter or fabG1g609a mutations. A higher resistance level of these combinations for these isolates would be indicative of possible new molecular interactions in katG and in INH resistance. Protein folding in combination with mutagenesis and enzymatic studies directly in Mtb would have to elucidate the specific molecular mechanisms and their effects on the resistance level.
Among isolates that harbor canonical mutations some interesting lineage patterns have emerged. These patterns are displayed in Supplementary Figure S5. First, the Indo-Oceanic group in our set displayed much greater tendency to only harbor an inhA promoter mutation as opposed to a katG mutation. 42% of Indo-Oceanic isolates belong to this group. The distant second was the Euro-American group, 5% of which harbored only an inhA promoter mutation. 68% of Euro-American group harbored a mutation in katG and one in inhA promoter. The distant second was the Beijing group in which 33% of the isolates harbored a mutation in both genes.
Codon 463 in katG has been shown not to be associated with resistance to INH and is commonly used to categorize clinical isolates into members of genetic groups 2 or 3, by a Arg 463 substitution, or genetic group 1, by a Leu 463 substitution.Citation60 As mentioned before, 58% of all resistant isolates and 63% of all susceptible isolates from four countries in our study harbored a R463L mutation and therefore belong to genetic group 1. Importantly, 19 of the 24 (or 79%) isolates harboring a novel mutation also harbored R463L and hence belonged to genetic group 1. The higher percentage of R463L among resistant isolates harboring a novel mutation (79% as compared to 58% for all resistant isolates from four countries or 59% for all resistant isolates from South Africa which is where most of the isolates with novel mutations came from) may indicate a higher tendency for genetic group 1 to take an uncommon evolutionary path and feature one of the rare resistance conferring mutations. This hypothesis is further supported by the fact that 14 of the 15 (93%) isolates that harbor fabG1g609a also harbored R463L. This would indicate more frequent evasion from molecular detection by genetic group 1 and lower sensitivity of molecular tests in regions of the world where genetic group 1 is more prevalent.
With the recent discovery of multiple additional resistance conferring mutations in katG by multiple investigators,Citation16,Citation64,Citation65,Citation66 it is a concern that a more widespread use of existing molecular diagnostics that miss these mutations might impose an artificial selection process where the mutants with canonical mutations are detected and eradicated through appropriate therapy but those with these novel variants go undetected and continue to spread. This may ultimately drive the Mtb INH-resistant population evolution from the current “hotspot” model (most mutations in a few loci in close proximity to each other) to more of a “whole-gene” convergence model. The scatter pattern of the novel mutations reported in this article suggests that a “multi-hotspot” model may also be a possibility. Much more representative data are needed for a more certain determination of these trends and emphasizes the need for global genomic surveillance of resistant phenotypes and for more flexible diagnostic platforms with the ability to interrogate broad regions of the genome.
As mentioned before, the remaining eight INHR isolates (2% of all INHR isolates) did not have katG, inhA promoter, or fabG1 mutations. We therefore explored the remainder of the genome looking for mutations previously associated with INH resistance that could potentially explain their phenotype. A subset of the “unexplained” 2% of INHR isolates were found to possess either an iniA H481Q or a fbpc 157292G-A mutation, both of which have been previously identified in INH-resistant Mtb isolates,Citation67 or an accD6 D229G mutation shown to play a role in the building blocks for de novo fatty acid biosynthesis by fatty acid synthase I (FAS I) that produce meromycolic acids.Citation68 We believe the latter is worthy of further investigation as mycolic acids are essential for the survival, virulence, and antibiotic resistance of the Mtb. It is clear that further work is needed to investigate and validate the true cause(s) of resistance for the remaining 2% of our isolates not containing the mutations traditionally associated with INH resistance.
Finally, our results have confirmed that some mono-INH resistance isolates do lack the canonical mutations and instead harbor novel mutations reported in this article. Patients infected with Mtb isolates harboring these mutations would go undetected by the currently available molecular tests for INH resistance and would be prime candidates for conversion to MDR-TB.
M. smegmatis is a convenient proxy for understanding functional consequences of mutations in Mtb because of its higher growth rate and its noninfectious nature. M. smegmatis, however, is not an accurate model for estimation of functional levels (e.g. resistance levels) in Mtb. As such, it is possible that mutations that do not cause (or cause low levels of) resistance in M. smegmatis, cause detectable and consequential levels of resistance in Mtb. For this reason, mutagenesis directly in Mtb, or at least in closer models such as H37ra would provide a more accurate functional-level estimation. In particular, the noncausative role of the two mutations R385W and D387G in INH resistance, as determined by M. smegmatis mutagenesis experiments, needs to be confirmed in Mtb or at least in a closer model such as H37ra.
Supplementary Figure S1
Download PDF (252.5 KB)Supplementary Figure S2
Download PDF (229.7 KB)Supplementary Figure S3
Download PDF (202.4 KB)Supplementary Figure S4
Download PDF (131.9 KB)Supplementary Figure S5
Download PDF (173.1 KB)Supplementary Table S1
Download PDF (24.5 KB)Supplementary Table S2
Download PDF (27 KB)Supplementary Table S3
Download PDF (27.9 KB)Supplementary Table S4
Download PDF (23.8 KB)Supplementary Table S5
Download PDF (70.8 KB)Supplementary Table S6
Download PDF (65 KB)Supplementary Table S7
Download PDF (26.2 KB)Supplementary Table S8
Download PDF (24.4 KB)Supplementary Table S9
Download PDF (25.3 KB)This work has been funded by National Institute of Allergy and Infectious Diseases (NIAID) grant NOs R01AI105185 and U01AI082229. Timothy C Rodwell was also supported by NIAID grant R01AI111435. The specimens for this project were provided by the Global Consortium for Drug-resistant TB Diagnostics (GCDD, http://gcdd.ucsd.edu). We thank Janice Kaping (UCSD, San Diego, CA, USA) for completing all the DST work. Thelma E Tupasi, Janice C Caoili, and Henry Evasco at the TDF and Kanchan Ajbani at the PDHNH helped collect and process the samples from Manila and India, respectively. Sanger sequencing was performed by the University of Hawaii under the supervision of James Douglas (University of Hawaii, Honolulu, HI, USA). Pyrosequencing was performed by Grace Lin under supervision of Edward Desmond at the California Department of Public Health, Microbial Diseases Laboratory, Richmond, CA, USA. Jessica N Torres, Afif Elghraoui, and Sarah M Ramirez-Busby have received scholarships from a grant (DUE-0966391) by the National Science Foundation.
Supplementary information for this article can be found on the Emerging Microbes & Infections' website (http://www.nature.com/EMI).
Notes
aIsolates that are INHR and RIFR and susceptible to the remaining five drugs are labeled as MDR. MDR+ are pre-XDR isolates that are INHR, RIFR, and resistant to at least a fluoroquinolone (FQ) or an aminoglycoside but not to both.
bDenotes polymorphisms whose role in causing resistance has been confirmed through mutagenesis.
cDenotes isolates that harbor katG R463L lineage marker.
dDenotes novel polymorphisms at previously reported positions.
eDenotes isolates that also harbor fabG1g609a.
fDenotes polymorphisms whose mutagenesis experiments were not successful. These are A139P and T394M.
gDenotes polymorphisms whose role in causing resistance has been disproved through mutagenesis. These are R385W and D387G.
aIsolates that are INHR and RIFR and susceptible to the remaining five drugs are labeled as MDR. MDR+ are pre-XDR isolates that are INHR, RIFR, and resistant to at least a fluoroquinolone (FQ) or an aminoglycoside but not to both. “Other Pre-XDR” isolates are those that are FQR and/or resistant to aminoglycoside drugs as well as to isoniazid but are susceptible to Rifampicin.
bDenotes isolates that harbor katG R463L lineage marker.
Related Research Data
- World Health Organization. Global Tuberculosis Report 2014.Geneva: WHO, 2014.Available at http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf (accessed 1 November 2014).
- World Health Organization.Global Tuberculosis Report 2013.Geneva: WHO, 2013.Available at http://www.who.int/tb/publications/global_report/en/ (accessed 3 July 2013).
- Boehme CC, Nabeta P, Hillemann D et al.Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med 2010; 363: 1005–1015.
- Ferro BE, García PK, Nieto LM, van Soolingen D.Predictive value of molecular drug resistance testing of Mycobacterium tuberculosis isolates in Valle del Cauca, Colombia. J Clin Microbiol 2013; 51: 2220–2224.
- Jacobson KR, Theron D, Victor TC, Streicher EM, Warren RM, Murray MB.Treatment outcomes of isoniazid-resistant tuberculosis patients, Western Cape Province, South Africa. Clin Infect Dis 2011; 53: 369–372.
- Varahram M, Nasiri MJ, Farnia P, Mozafari M, Velayati AA.A retrospective analysis of isoniazid-monoresistant tuberculosis: among Iranian pulmonary tuberculosis patients. Open Microbiol J 2013; 8: 1–5.
- Gagneux S, Burgos M V, DeRiemer K et al.Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLoS Pathog 2006; 2: e61.
- Yu S, Girotto S, Lee C, Magliozzo RS.Reduced affinity for Isoniazid in the S315T mutant of Mycobacterium tuberculosis KatG is a key factor in antibiotic resistance. J Biol Chem 2003; 278: 14769–14775.
- Pym AS, Saint-Joanis B, Cole ST.Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect Immun 2002; 70: 4955–4960.
- Guo H, Seet Q, Denkin S, Parsons L, Zhang Y.Molecular characterization of isoniazid-resistant clinical isolates of Mycobacterium tuberculosis from the USA. J Med Microbiol 2006; 55: 1527–1531.
- Larsen MH, Vilchèze C, Kremer L et al.Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol Microbiol 2002; 46: 453–466.
- Rodwell TC, Valafar F, Douglas J et al.Predicting extensively drug-resistant Mycobacterium tuberculosis phenotypes with genetic mutations. J Clin Microbiol 2014; 52: 781–789.
- Wei C-J, Lei B, Musser JM, Tu S-C.Isoniazid activation defects in recombinant Mycobacterium tuberculosis catalase-peroxidase (KatG) mutants evident in InhA inhibitor production. Antimicrob Agents Chemother 2003; 47: 670–675.
- Siqueira HR, Freitas FA, Oliveira DN, Barreto AM, Dalcolmo MP, Albano RM.Isoniazid-resistant Mycobacterium tuberculosis strains arising from mutations in two different regions of the katG gene. J Bras Pneumol 2009; 35: 773–779.
- Ramaswamy SV, Reich R, Dou SJ et al.Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2003; 47: 1241–1250.
- Ando H, Miyoshi-Akiyama T, Watanabe S, Kirikae T.A silent mutation in mabA confers isoniazid resistance on Mycobacterium tuberculosis. Mol Microbiol 2014; 91: 538–547.
- Hillery N, Groessl EJ, Trollip A et al.The Global Consortium for Drug-resistant Tuberculosis Diagnostics (GCDD): design of a multi-site, head-to-head study of three rapid tests to detect extensively drug-resistant tuberculosis. Trials 2014; 15: 434.
- Canetti G, Fox W, Khomenko A et al.Advances in techniques of testing mycobacterial drug sensitivity, and the use of sensitivity tests in tuberculosis control programmes. Bull World Health Organ 1969; 41: 21–43.
- Garfein RS, Catanzaro DG, Rodwell TC et al.Phenotypic and genotypic diversity in a multinational sample of drug-resistant Mycobacterium Tuberculosis isolates. Int J Tuberc Lung Dis 2015; 19: 420–427.
- Kamerbeek J, Schouls L, Kolk A et al.Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol 1997; 35: 907–914.
- Cowan LS, Diem L, Monson T et al.Evaluation of a two-step approach for large-scale, prospective genotyping of Mycobacterium tuberculosis isolates in the United States. J Clin Microbiol 2005; 43: 688–695.
- Mazars E, Lesjean S, Banuls AL et al.High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci U S A 2001; 98: 1901–1906.
- Shabbeer A, Cowan LS, Ozcaglar C et al.TB-Lineage: an online tool for classification and analysis of strains of Mycobacterium tuberculosis complex. Infect Genet Evol 2012; 12: 789–797.
- Aminian M, Couvin D, Shabbeer A et al.Predicting Mycobacterium tuberculosis complex clades using knowledge-based Bayesian networks. Biomed Res Int 2014; 2014: 398484.
- Ajbani K, Lin SY, Rodrigues C et al.Evaluation of Pyrosequencing for Detecting Extensively Drug-resistant Tuberculosis (XDR-TB) in clinical isolates from four high-burden countries. Antimicrob Agents Chemother 2015; 59: 414–420.
- Van Helden PD, Victor TC, Warren RM, van Helden EG.Isolation of DNA from Mycobacterium tubercolosis. Methods Mol Med 2001; 54: 19–30.
- QIAGEN. QIAGEN Genomic DNA Handbook 04/2012.Hilden: QIAGEN Technical Support, 2012.
- Van der Auwera GA, Carneiro MO, Hartl C et al.From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatis 2013; 11: 11.10.1–11.10.33.
- McKenna A, Hanna M, Banks E et al.The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
- DePristo MA, Banks E, Poplin R et al.A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
- Li H, Handsaker B, Wysoker A et al.The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
- Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT.BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011; 27: 1691–1692.
- Koboldt DC, Chen K, Wylie T et al.VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009; 25: 2283–2285.
- Koboldt DC, Zhang Q, Larson DE et al.VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22: 568–576.
- Lin SY, Rodwell TC, Victor TC et al.Pyrosequencing for rapid detection of extensively drug-resistant Mycobacterium tuberculosis in clinical isolates and clinical specimens. J Clin Microbiol 2014; 52: 475–482.
- Borrell S, Teo Y, Giardina F et al.Epistasis between antibiotic resistance mutations drives the evolution of extensively drug-resistant tuberculosis. Evol Med public Heal 2013; 2013: 65–74.
- Koch A, Mizrahi V, Warner DF.The impact of drug resistance on Mycobacterium tuberculosis physiology: what can we learn from rifampicin? Emerg Microbes Infect 2014; 3: e17.
- Xu X, Vilchèze C, Av-Gay Y, Gómez-Velasco A, Jacobs WR.Precise null deletion mutations of the mycothiol synthesis genes reveal their role in isoniazid and ethionamide resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 2011; 55: 3133–3139.
- Zhang Y, Heym B, Allen B, Young D, Cole S.The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992; 358: 591–593.
- Shcherbakov D, Akbergenov R, Matt T, Sander P, Andersson DI, Böttger EC.Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in-vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol 2010; 77: 830–840.
- Parish T, Roberts G, Laval F, Schaeffer M, Daffé M, Duncan K.Functional complementation of the essential gene fabG1 of Mycobacterium tuberculosis by Mycobacterium smegmatis fabG but not Escherichia coli fabG. J Bacteriol 2007; 189: 3721–3728.
- Burian J, Yim G, Hsing M et al.The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Res 2013; 41: 10062–10076.
- Karunakaran P, Davies J.Genetic antagonism and hypermutability in Mycobacterium smegmatis. J Bacteriol 2000; 182: 3331–3335.
- Johnson R, Streicher EM, Louw GE, Warren RM, van Helden PD, Victor TC.Drug resistance in Mycobacterium tuberculosis. Curr Issues Mol Biol 2006; 8: 97–111.
- Parish T, Stoker NG.Use of flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 2000; 146: 1969–1975.
- Thermo Scientific.Phusion Polymerase.Waltham, MA: Thermo Scientific, 2014.Available at http://www.thermoscientificbio.com/pcr-enzymes-master-mixes-and-reagents/phusion-high-fidelity-dna-polymerase/ (accessed 1 May 2014).
- Edelheit O, Hanukoglu A, Hanukoglu I.Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol 2009; 9: 61.
- Chan RC, Hui M, Chan EWC et al.Genetic and phenotypic characterization of drug-resistant Mycobacterium tuberculosis isolates in Hong Kong. J Antimicrob Chemother 2007; 59: 866–873.
- Heym B, Alzari PM, Honore N, Cole ST.Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol 1995; 15: 235–245.
- Hillemann D, Weizenegger M, Kubica T, Richter E, Niemann S.Use of the genotype MTBDR assay for rapid detection of rifampin and isoniazid resistance in Mycobacterium tuberculosis complex isolates. J Clin Microbiol 2005; 43: 3699–3703.
- Choi JH, Lee KW, Kang HR et al.Clinical efficacy of direct DNA sequencing analysis on sputum specimens for early detection of drug-resistant Mycobacterium tuberculosis in a clinical setting. Chest 2010; 137: 393–400.
- Jagielski T, Grzeszczuk M, Kamiński M et al.Identification and analysis of mutations in the katG gene in multidrug-resistant Mycobacterium tuberculosis clinical isolates. Pneumonol Alergol Pol 2013; 81: 298–307.
- Zaker Bostanabad S, Titov LP, Slizen VV, Taghikhani M, Bahrmand A.katG mutations in isoniazid-resistant strains of Mycobacterium tuberculosis isolates from Belarusian patients. Tuberk Toraks 2007; 55: 231–237.
- Rouse DA, Li Z, Bai GH, Morris SL.Characterization of the katG and inhA genes of isoniazid-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 1995; 39: 2472–2477.
- Vilchèze C, Wang F, Arai M et al.Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med 2006; 12: 1027–1029.
- Pretorius GS, van Helden PD, Sirgel F, Eisenach KD, Victor TC.Mutations in katG gene sequences in isoniazid-resistant clinical isolates of Mycobacterium tuberculosis are rare. Antimicrob Agents Chemother 1995; 39: 2276–2281.
- Fenner L, Egger M, Bodmer T et al.Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2012; 56: 3047–3053.
- Cockerill FR 3rd, Uhl JR, Temesgen Z et al.Rapid identification of a point mutation of the Mycobacterium tuberculosis catalase-peroxidase (katG) gene associated with isoniazid resistance. J Infect Dis 1995; 171: 240–245.
- Ramaswamy S, Musser JM.Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis 1998; 79: 3–29.
- Sreevatsan S, Pan X, Stockbauer KE et al.Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci USA 1997; 94: 9869–9874.
- Tukvadze N, Kempker RR, Kalandadze I et al.Use of a molecular diagnostic test in AFB smear positive tuberculosis suspects greatly reduces time to detection of multidrug resistant tuberculosis. PLoS One 2012; 7: e31563.
- García-Sierra N, Lacoma A, Prat C et al.Pyrosequencing for rapid molecular detection of rifampin and isoniazid resistance in Mycobacterium tuberculosis strains and clinical specimens. J Clin Microbiol 2011; 49: 3683–3686.
- Piatek AS, Telenti A, Murray MR et al.Genotypic analysis of Mycobacterium tuberculosis in two distinct populations using molecular beacons: implications for rapid susceptibility testing. Antimicrob Agents Chemother 2000; 44: 103–110.
- Ramasubban G, Therese KL, Lakshmipathy D, Sridhar R, Meenakshi N, Madhavan HN.Detection of novel and reported mutations in the rpoB, katG and inhA genes in multidrug-resistant tuberculosis isolates: a hospital-based study. J Glob Antimicrob Resist 2014; 3: 1–4.
- Ando H, Kondo Y, Suetake T et al.Identification of katG mutations associated with high-level isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2010; 54: 1793–1799.
- Syah YM, Retnoningrum DS, Noer AS, Shigeoka S, Natalia D.Novel mutations in katG gene of a clinical isolate of isoniazid-resistant Mycobacterium tuberculosis. Biologia (Bratisl) 2012; 67: 41–47.
- Lee AS, Lim IH, Tang LL, Telenti A, Wong SY.Contribution of kasA analysis to detection of isoniazid-resistant Mycobacterium tuberculosis in Singapore. Antimicrob Agents Chemother 1999; 43: 2087–2089.
- Daniel J, Oh TJ, Lee CM, Kolattukudy PE.AccD6, a member of the Fas II locus, is a functional carboxyltransferase subunit of the acyl-coenzyme A carboxylase in Mycobacterium tuberculosis. J Bacteriol 2007; 189: 911–917.