
Abstract
Humoral responses are essential for the protective efficacy of most Ebola virus (EBOV) candidate vaccines; however, the in vivo development of protective anti-EBOV B-cell responses is poorly defined. Here, by using the virus-like particle (VLP) as a model antigen, we demonstrate that humoral responses are generated through follicular B-cell and T-cell-dependent mechanisms in a mouse model of EBOV infection. In addition, we show that the inclusion of the clinical-grade dsRNA adjuvant known as poly-ICLC in VLP vaccinations both augments and sustains germinal center B-cell reactions, antigen-specific B-cell frequencies and anti-EBOV serum titers. Finally, we used mice that were deficient in either B-cells or T-cell-dependent antibody production to distinguish the contributing roles of EBOV humoral responses. We demonstrate that while anti-EBOV antibody responses promote protection, VLP-vaccinated mice can survive EBOV infection in the absence of detectable anti-EBOV antibodies. Moreover, we found that adjuvant signaling could circumvent the complete requirement for B-cell immunity in protection against EBOV. Collectively, these studies may prove valuable for the characterization and future development of additional EBOV vaccine candidates.
Introduction
The 2013–2016 Ebola virus disease (EVD) epidemic in Western Africa, which involved more than 28 000 individuals and claimed more than 11 000 lives (World Health Organization, http://www.who.int/en/; Centers for Disease Control and Prevention, http://www.cdc.gov/), triggered the advancement of several medical countermeasures against the Ebola virus (EBOV), some of which had been in development for decades.Citation1, Citation2, Citation3 Multiple vaccine platforms expressing or consisting of the EBOV trimeric glycoprotein (GP1,2) have been generated, and several are currently under clinical evaluation.Citation4, Citation5, Citation6, Citation7, Citation8 However, there are fundamental gaps in our understanding of how candidate vaccines induce protective host responses, and a definitive immune correlate for vaccine efficacy has yet to be defined.Citation9, Citation10
On the basis of studies using nonhuman primate (NHP) and rodent models of EVD, achieving both cellular and humoral responses against EBOV is thought to be critical for protection.Citation11, Citation12 The roles of EBOV-specific CD4+ and CD8+ T-cell responses have been extensively characterized; nonetheless, they remain complex and appear to be largely vaccine platform-dependent. Adenovirus 5 and Venezuelan equine encephalitis virus-like replicon-based EBOV vaccine studies have demonstrated a critical role for CD8+ T-cell responses in protecting against NHP and murine EBOV infection.Citation13, Citation14, Citation15 However, dispensable roles for EBOV-specific CD8+ T-cell responses have been supported by studies using recombinant vesicular stomatitis virus and adenovirus Hu5 vaccine platforms.Citation16, Citation17, Citation18 Murine studies from our group that used EBOV viral-like particles (VLPs) have shown a similar importance for T-cells; however, adjuvants have impacted the establishment and relative contributions of these responses in protecting against EVD.Citation19, Citation20, Citation21 Despite reported differences and alternative mechanisms for T-cell-mediated immunity between EBOV-based vaccine platforms, most converge on an obligate requirement for humoral responses.Citation16, Citation17, Citation18, Citation21, Citation22 Further supporting this importance is the preclinical success of anti-EBOV antibodies (for example, ZMapp, ZMab) in treating EBOV infection.Citation23, Citation24, Citation25
The production of antibodies following a direct interaction between B-cells and the cognate antigen can occur through T-cell-independent mechanisms (TI); however, the generation of high-affinity class-switched antibodies is dependent on follicular T-cell help (TFH). The formation of T-cell-dependent (TD) antibody responses can be shaped through several key events including (i) the induction of germinal center (GC) B-cell reactions; (ii) TFH quantity and quality; (iii) activation-induced cytidine deaminase (AICDA/AID)-mediated somatic hypermutation and isotype class-switching; and (iv) the selection and differentiation of GC B-cells to form the antigen-specific B-cell compartment.Citation26, Citation27 However, in vivo studies that define how EBOV B-cell immunity is established, the relative contributions of TI and TD B-cell mechanisms and the direct requirement for B-cell responses to protect against EBOV infection have been limited to date.Citation17, Citation28
Previously, we demonstrated that the vaccination of laboratory mice, guinea pigs, or NHPs with a VLP consisting of EBOV matrix protein (VP-40) and GP1,2 elicited complete protection against EBOV infection.Citation29, Citation30, Citation31 We also discovered that the inclusion of the clinical-grade dsRNA polyinosinic-polycytidylic acid (poly-IC) derivative known as poly-IC poly-l-lysine carboxymethylcellulose (poly-ICLC) in VLP vaccine preparations increased EBOV GP1,2-specific antibody titers and durable protection from EVD in mice.Citation19, Citation20 Here, we use VLP as a model system to examine the establishment and requirement for EBOV B-cell immunity in mice and the impact of poly-ICLC adjuvant signaling on VLP-mediated B-cell responses.
MATERIALS AND METHODS
Reagents
Poly-ICLC (Hiltonol) was provided by Oncovir (Washington, DC, USA). Ebola VLPs were produced as previously described.Citation20, Citation30 In brief, 293T cells were transfected with Ebola Zaire (Kikwit) virus glycoprotein and VP-40. VLP supernatants were collect at 72 h post-transfection, they were sucrose gradient-purified, and their total protein content was determined using a bicinchoninic acid protein assay. To ensure sterility, the VLPs were irradiated at 1e6 rad, and they contained less than 25 EU/mL endotoxin and less than 10 colony-forming units (CFU) of bacteria per vaccination. The particular lot of VLP within this study was previously used for EBOV vaccine studies and has been extensively characterized.Citation19, Citation32 The GP content for these studies was determined by Western blot and fixed at a 10 μg GP dose for the vaccinations. The VLPs were maintained at −80 °C and diluted in sterile saline and/or combined with poly-ICLC prior to vaccination.
Mouse strain and vaccinations
C57BL/6 (NCI Charles River Strain Code 027, Jackson Stock No. 000664), CD40-deficient (Jackson Stock No. 002928), μMT (Jackson Stock No. 002288) and AID-deficient mice (kind gifts from Drs Pat Gearhart, Robert Maul, NIAIA, Baltimore and Rafael Casellas, NIH, Bethesda) were each vaccinated intramuscularly with VLP (10 μg GP1,2 content) or VLP (10 μg GP1,2 content) plus 10 μg of poly-ICLC at 3-week intervals (day 0, day 21).
Enzyme-linked immunosorbent assays
ELISAs were performed as previously described.Citation19 In brief, blood was collected from the vaccinated mice at the indicated time points in Vacutainer serum-separating tubes. ELISA plates were coated with recombinant mammalian cell-expressed EBOV GP1,2 at 2 μg/mL in phosphate-buffered saline (PBS; Corning Life Sciences, Corning, NY, USA). The sera were diluted by half-log dilutions starting at 1:100, and then incubated for 1 h on GP1,2-coated plates. The plates were washed and then incubated with the indicated secondary horseradish peroxidase (HRP) antibody. ELISAs were developed using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate/stop solution and measured on a Tecan plate reader. The absorbance cut-off was determined as the background+0.2 O.D.
Flow cytometry
Single-cell suspensions of draining lymph nodes and spleens were collected at the indicated time points. The cells were washed using FACS buffer (PBS, 0.5% BSA and 2 mM EDTA; Corning, Sigma, St Louis, MO, USA), lysed with red blood cell (RBC) buffer (Sigma), and subsequently counter-stained. The B-cell staining included B220 (Becton-Dickinson Biosciences (BD), Franklin Lanes, NJ, USA Clone RA3-6B2), IgM (BD Clone R6-60.2), IgD (eBioscience, San Diego, CA, USA Clone 11-26C), CD38 (BD Clone 90), CD95 (BD Clone Jo-2), and T & B Cell Activation Antigen (BD Clone GL-7). T follicular helper cell staining was performed by a primary incubation with CXCR5 (BD Clone 2G8) followed by secondary incubation using goat anti-rat (H+L)-biotin (Jackson ImmunoResearch, West Grove, PA, USA 112-067-003). Subsequently, the cells were counter-stained with streptavidin (BD 557598), CD3 (BD Clone 500A2), CD4 (BD Clone RM4-5), PD-1 (eBioscience Clone RMP1-30), and ICOS (BD Clone 7E.17G9). All the samples were Fc-blocked (anti-CD16/CD32, BD) and stained to evaluate their viability (live/dead aqua, Invitrogen, Carlsbad, CA, USA) before counter-staining. The data were collected on a BD FACSCanto II or BD FACSAria II and analyzed using FlowJo (Treestar, Ashland, OR, USA).
Neutralization assays
Neutralizing antibody titers from the serum samples were determined using recombinant vesicular stomatitis Indiana virus (rVSV) particles that coexpressed EBOV GP1,2 and enhanced green fluorescent protein (eGFP) (a kind gift from Kartik Chandran, Albert Einstein College of Medicine). In brief, serum dilutions beginning at 1:10 were incubated at 37 °C for 1 h with rVSV and 5% v/v Hemo-lo guinea pig complement (Cedarlane Laboratories, Burlington, NC, USA). rVSV/serum complexes were then incubated with 1 × 105 FreeStyle 293F cells (ThermoFisher Scientific, Waltham, MA, USA) at 1 × 106 cells/mL for 18–20 h at 37 °C. The infection percentage, as determined by eGFP expression, was measured using fluorescence-activated cell sorting (FACS). The data were collected on a BD Canto II and LSR II. The neutralization was calculated by normalizing the infection percentages to the infections performed in the presence of control sera from non-vaccinated laboratory mice.
B-cell ELISPOTs
ELISPOTs were performed according to the MabTech (Cincinnati, OH, USA) protocol. In brief, cryopreserved splenocyte populations were thawed, washed 2 × with complete medium, and adjusted to 2 × 106 cells/mL. Splenocyte suspensions (0.1 mL) were added to recombinant EBOV GP1,2-coated ELISPOT plates and then incubated overnight at 37 °C with 95/5 oxygen/CO2. The plates were then washed, incubated with biotin-IgG secondary antibody (MabTech) followed by streptavidin-HRP (MabTech) and developed using TMB substrate/stop solution. Imaging/counting was performed using a CTL Immunospot instrument (Cellular Technology, Shaker Heights, OH, USA).
Ebola virus infections
Laboratory mice were inoculated intraperitoneally with a target dose of 1000 pfu of mouse-adapted Ebola virus/H. sapiens-tc/COD/1976/Yambuku-Mayinga (ma-EBOV) on day 28 after the last vaccination or during secondary inoculations. All the live-virus studies were conducted under maximum (biosafety level 4) containment. Clinical observations were recorded throughout the study starting on the day of virus inoculation. Moribund mice were euthanized according to institution-approved clinical scoring.
Statistical analysis
All the statistical analyses were performed using GraphPad Prism (Windows V6). Data comparisons using unpaired parametric Student’s t-tests were performed for titer analysis and immune populations. A survival curve comparison was performed using a log-rank (Mantel–Cox) test.
RESULTS
VLP vaccination induces robust germinal center B-cell responses, which are augmented with the inclusion of poly-ICLC
Prior VLP vaccination studies have demonstrated the induction of EBOV GP1,2, antibody responses and that these responses can be heightened with the inclusion of adjuvants. Recently, we demonstrated that the dsRNA adjuvant poly-ICLC provided enhanced VLP-mediated anti-EBOV titers and influenced the antibody-isotype.Citation19, Citation20 The production of high-affinity class-switched antibodies is dependent on germinal center (GC) formation;Citation26, Citation33 however, limited characterization of this B-cell compartment has been reported for vaccine platforms that were developed against EBOV.Citation28 Therefore, we examined if VLP vaccination resulted in the generation of GC reactions and if poly-ICLC would impact these responses. Mice were vaccinated with VLP (intramuscularly) in the presence or absence of poly-ICLC using a prime-boost schedule at three-week intervals (day 0, day 21). VLP-vaccinated mice were characterized by an approximate three-fold increase in draining lymph node (dLN, popliteal) cellularity at day 10 subsequent to prime vaccination, whereas the inclusion of poly-ICLC resulted in an approximate seven-fold increase (Figure ). However, the relative frequency of B220+ B-cells was unaltered (Figure ). On day 10, subsequent to prime vaccination, GC B-cells (B220+GL7+CD95+) were clearly identified within the dLN of both VLP-vaccinated groups. However, the relative frequencies and total numbers of GC B-cells were increased in the presence of poly-ICLC (Figure ).
We next determined the impact of vaccine boosting on VLP-mediated GC reactions. As with prime vaccination, GC B-cells were detected within the dLN on day 28 (day 7 post-boosting), and, consistent with the prime vaccination results, the inclusion of the adjuvant significantly augmented the relative frequency of GC B-cells (Figure , left panel). Additional phenotypic characterization of the B-cell compartment supported the induction of activated class-switched (B220+GL7+CD95+CD38lowIgD−IgM−) B-cells following VLP vaccination with heightened frequencies observed when adding poly-ICLC (Figure , right panel). Notably, the relative frequency of GC B-cells increased after boosting in the presence of poly-ICLC, whereas VLP-only-vaccinated mice presented comparable GC B-cell frequencies following both prime and boost vaccinations (Figures ). VLP-mediated GC reactions were still present within the dLN at four weeks post-boosting (day 49) with a continued increase in GC B-cell frequencies being observed in the presence of the adjuvant (Figure ). Consistent with GC B-cell dynamics, the VLP-induced antibody responses plateaued following vaccination in the absence of the adjuvant. However, the inclusion of poly-ICLC resulted in a continual rise of total anti-GP1,2 IgG titers and enhanced EBOV neutralization up to one month post-final vaccination (day 49), a time-point associated with acute protection from EBOV infection (Supplementary Figures S1A and S1C). Durable (>5 months post-vaccination) anti-EBOV GP1,2 IgG responses were additionally augmented in the presence of poly-ICLC (Supplementary Figure S1B, top). Interestingly, after one, two or three vaccinations, the EBOV GP1,2-specific IgM responses were marginal and were detected only in the presence of the adjuvant (Supplementary Figure S1B, bottom).
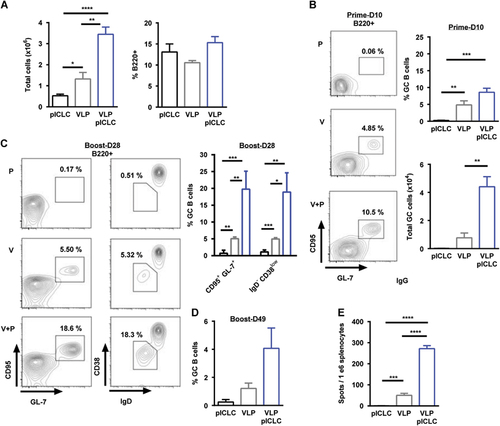
Because productive GC reactions result in the generation of antibody-secreting cells (ASCs), we measured the frequency of EBOV GP1,2-specific B-cells. Antigen-specific B-cells could not be detected after prime vaccination (data not shown), but we observed an approximate five-fold increase in ASC frequencies after the inclusion of poly-ICLC following boosting (Figure ). GP1,2-specific B-cells were still detectable up to day 49 in the presence of adjuvant, but they approached our lower limit of detection for VLP vaccination alone (data not shown). Altogether, we demonstrate that VLP-mediated B-cell responses are associated with GC formation and that the addition of poly-ICLC as an adjuvant resulted in augmented GC B-cell frequencies, increased generation of EBOV GP1,2-specific ASCs and elevated antibody titers.
VLP-mediated humoral immunity is established through follicular B-cell and T-cell-dependent mechanisms, but it is partially dispensable for protection
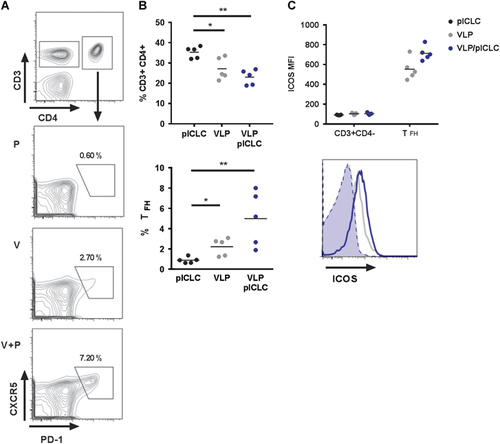
The quality and quantity of GC B-cell formation is dependent on TFH cells.Citation34 Consistent with our recent findings and the relationship between TFH and the formation of GC reactions,Citation19, Citation35 VLP-mediated TFH (CD3+CD4+PD-1+CXCR5hi) cells are generated during vaccination (Figures ). Moreover, the relative size of the TFH compartment is increased with the addition of adjuvant, suggesting that poly-ICLC augments humoral immunity by promoting TFH and GC B-cell reactions (Figures ). Consistent with the cellular phenotyping, high levels of inducible T-cell costimulator (ICOS, CD278) was detected on TFH cells following vaccination with a subtle increase of expression after adjuvant inclusion (Figure ). Interestingly, the relative frequency of dLN CD3+CD4+ T-cells declined subsequent to vaccination (Figure ).
CD40-CD40L interactions between GC B-cells and TFH cells are required to induce productive T-cell-dependent (TD) antibody responses.Citation36 We therefore determined the direct in vivo contribution of this pathway during VLP vaccination. The tumor necrosis factor superfamily, receptor 5 knockout (CD40−/−) mice have defective TD humoral immune responses while also being capable of establishing TI antibody responses.Citation37 Wild-type and CD40−/− mice were prime-boost-vaccinated (day 0, day 21) with VLP and EBOV GP1,2-specific antibody titers were analyzed on day 35. Wild-type mice that were vaccinated with VLP displayed robust IgG antibody titers; however, the CD40−/− mice had no detectable anti-GP1,2-specific IgG or IgM responses (Figure and Supplementary Figure S2A). The VLP candidate vaccine consists of both EBOV GP and VP-40; we therefore also determined antibody reactivity against VP-40. Consistent with the lack of GP1,2 antibody responses, the CD40−/− VLP-vaccinated mice also failed to establish anti-VP-40 humoral responses (Supplementary Figure S2B).
Previous in vivo characterizations of EBOV candidate vaccines have suggested an obligate requirement for anti-EBOV antibodies for efficacy.Citation16, Citation17, Citation21, Citation22 We therefore tested the protective contribution of humoral immunity using CD40−/− mice, which displayed a lack of VLP-specific antibody responses. The prime-boost vaccination of wild-type mice with VLP or VLP/poly-ICLC resulted in acute protection against a typically lethal dose of EBOV (Figure ). Surprisingly, we also observed partial protection in VLP-vaccinated CD40−/− mice (Figure ). In two separate studies, the control mice succumbed to EBOV infection by day 10, whereas a combined 60% (n=24/40) of the VLP-vaccinated mice were protected from disease in the absence of detectable EBOV GP1,2-specific antibody responses. Importantly, we observed similar morbidity within unvaccinated wild-type (mean survival= 7 days) and CD40−/− mice (mean survival=6.5 days; P-value= 0.62), suggesting comparable EBOV lethality across murine strains. Poly-ICLC did not significantly alter the protection in VLP-vaccinated CD40−/− mice (Figure ).
We speculated that despite the lack of TI responses upon VLP vaccination, EBOV infection may result in the generation of protective low-affinity antibody responses. To test this hypothesis, we analyzed the antibody responses following EBOV infection in the mice that had survived EBOV infection. All the surviving wild-type mice had robust IgG titers against EBOV GP1,2, whereas the CD40−/− mice had a near complete absence of antibody responses, even subsequent to EBOV infection (Figure ). Of the 14 CD40−/− survivors analyzed after EBOV infection, only 3 had marginal anti-EBOV GP1,2 IgG titers and none mounted detectable IgM responses (Figure ). On day 28 subsequent to primary EBOV infection, the mice were once again inoculated with a typically lethal dose of EBOV. Consistent with the initial survival results, all the mice were protected from secondary EBOV infection, including those mice without detectable anti-GP1,2 antibody responses (Figure ). Altogether, these results support the idea that EBOV-specific antibody responses following either VLP vaccination or EBOV infection are generated through follicular B-cell and TD mechanisms. However, protection from EBOV lethality could be achieved in the absence of these TD humoral immune responses.
Protection from EBOV infection in the absence of antibody affinity maturation or class-switched humoral responses
Our finding in which mice survived a typical lethal EBOV inoculation in the absence of anti-EBOV GP1,2 antibodies is contrary to previous reports. To confirm our results, we used activation-induced cytidine deaminase-deficient (AICDA/AID−/−) mice that fail to generate high-affinity IgG isotype antibodies.Citation38 Wild-type or AID−/− mice were prime-boost-vaccinated with VLP as described above. VLP vaccination of wild-type mice generated robust IgG responses; however, AID−/− mice were characterized by a complete absence of class-switched EBOV GP1,2-specific IgG titers (Figure ). To determine the protective role of these IgG responses, wild-type and AID−/− mice were inoculated with a typically lethal dose of EBOV. In accordance with our CD40−/− studies, we observed ~60% protection against EBOV infection in mice lacking IgG antibody responses (Figure ). Moreover, wild-type and AID−/− control mice succumbed to EBOV infection at similar rates (wild-type mean survival=7 days, AID−/− mean survival=7.5 days; P=0.44). VLP-vaccinated AID−/− mice mounted marginal IgM responses that were not seen in wild-type mice; these responses do not appear to be required for protection (Figure ).
Adjuvant-enhanced protection against EBOV infection in the complete absence of B-cells
Despite the agreement of our CD40−/− and AID−/− mouse study results, previous experiments utilizing B-cell-deficient mice (for example, μMT, Jh−/−) have suggested that vaccine-induced antibody responses are obligatory for protection against EBOV.Citation12, Citation17, Citation21 However, μMT and Jh−/− mice have a developmental block in B-cell lymphopoiesis and therefore display a complete loss of the B-cell compartment.Citation39 Indeed, we observed that VLP-vaccinated μMT mice failed to establish EBOV GP1,2-specific antibody responses and were not protected from EBOV infection (Figures ). Consistent with previous reports, in the absence of B-cells, VLP vaccination alone failed to protect mice from a typically lethal dose of EBOV. However, surprisingly, the inclusion of poly-ICLC during μMT VLP vaccination rescued the protection of the mice from EBOV lethality despite the complete absence of B-cells (Figure ). This finding suggests that adjuvant signaling is inducing a B-cell-independent mechanism to protect against EBOV infection.
DISCUSSION
The mechanism of protection established by EBOV vaccine platforms has been much-debated, with no definitive correlate identified to date. As EBOV candidate vaccines progress towards Food and Drug Administration (FDA) licensure in the United States, increased focus on identifying correlates of immune protection will be required. Moreover, the potential lack of human efficacy data from EBOV vaccine trials may require licensure in the United States via the FDA ‘Animal Rule’.Citation10, Citation40 Therefore, our ability to interpret EBOV vaccine responses accurately depends upon well-defined animal models.
The ma-EBOV model has been critical for the characterization of protective Ebola vaccines and therapeutics.Citation41, Citation42 In addition, the ability to conduct studies in the context of wild-type and knockout (for example, Jh4 −/−, β2−/−, CD4−/−, CD8−/−, CD11−/−) mouse models has provided insight into the mechanisms of protection against EBOV infection.Citation18, Citation21, Citation43, Citation44 Previous studies have demonstrated the critical importance of anti-EBOV antibody vaccine responses.Citation16, Citation17, Citation22 The preclinical success of EBOV antibody therapeutics additionally supports an essential role for humoral immunity in the protection against EVD.Citation23, Citation24, Citation45 However, little is known about the early EBOV B-cell initiating events that are required for the establishment of these responses.Citation28
Within this study, we show that the EBOV GP1,2-specific humoral responses that were induced by both VLP vaccination and EBOV infection are predominately established through T-cell-dependent mechanisms. We find that VLP vaccinations generate robust and sustained GC B-cell reactions that are detectable at least four weeks post-vaccination, and these responses were augmented through dsRNA signaling as provided by poly-ICLC. Of interest, GC B-cell responses were not detectable following vaccination with equal amounts of soluble recombinant EBOV GP1,2, suggesting differences in GC formation associated with the display of EBOV GP1,2 (data not shown). Finally, our studies indicated that antibody responses promote protection against typically lethal EBOV infection, but they are neither obligate nor predictive of survival.
Our finding of protection from Ebola lethality in the absence of EBOV-specific antibodies builds upon the results of previous reports.Citation16, Citation17, Citation22 Prior murine studies that defined an obligate role for antibody-mediated protection against EVD were obtained from knockout models with a complete loss of the B-cell compartment.Citation16, Citation21 These studies demonstrated that B-cell-deficient mice were not protected following EBOV vaccination, which is consistent with our findings. Immune depletion studies within NHP models have additionally been used to delineate the requirement for B-cells in vaccine-induced EBOV immunity, and previous EBOV rVSV vaccine platform studies within NHPs demonstrated a critical role for CD4+ T-cells in the establishment of anti-GP1,2 antibody responses.Citation17 Our data support a similar mechanism within mice and extend this observation by linking the requirement for CD40/CD40L interaction.
However, contrary to previous reports, our studies demonstrate that an absence of antibodies does not result in a complete loss of protection from EVD. The animal models (CD40−/− and AID−/−) used within this study have no known defects in T or B-cell development and are fully capable of producing T-cell-independent antibodies. We observed ~60% protection against EBOV infection in both murine models following VLP vaccination, despite a failure to produce anti-EBOV GP1,2-specific antibodies. Moreover, in support of antibody-independent protection, we found that the inclusion of poly-ICLC in VLP preparations could partially rescue protection against EBOV lethality, even in animals which have a complete loss of B-cells. Understanding the mechanism by which this is achieved will be the focus of future studies.
Our results support the idea that anti-EBOV GP1,2-specific antibody responses are generated via follicular B-cell and TD mechanisms. Although we failed to detect TI antibody responses following either VLP vaccination or EBOV infection, we cannot preclude contributions from low-affinity antibodies that were unmeasurable within our studies. However, our finding of poly-ICLC-mediated protection in VLP-vaccinated B-cell-deficient mice would suggest a minor, if any, contribution by such mechanisms within our model. Previously, we demonstrated that EBOV-specific T-cell responses following VLP vaccination are essential for protection.Citation19, Citation20, Citation21 Prior studies using the VLP as a pre- or post-exposure therapeutic agent have reported a role for innate immune components in protecting against EVD.Citation43, Citation46 Therefore, these T-cell or innate immune responses may provide unknown compensatory mechanisms in protecting against EBOV lethality in the absence of anti-EBOV antibodies. Questions about the mechanisms of VLP efficacy in the absence of anti-EBOV antibodies are currently being investigated.
In opposition to an obligate effort by humoral components in protecting against EBOV lethality, we illustrate a division of labor for VLP-mediated anti-EBOV immunity. The finding of a loss of VLP-mediated efficacy in B-cell-deficient mice, yet protection without detectable anti-EBOV antibodies, potentially suggests an unappreciated role for antibody-independent B-cell mechanisms in promoting protection.Citation47, Citation48 The additional finding that the requirement for B-cells could be bypassed with the inclusion of dsRNA signaling suggests further unknown redundancies or compensatory mechanisms in the protection against EBOV lethality. Together, the data in this work provide a comprehensive description of the establishment and requirement of VLP-mediated EBOV humoral immunity. Moreover, they highlight intriguing gaps in our knowledge of less conventional roles for B-cell immune functions.
Supplementary Figure S1
Download PDF (203.7 KB)Supplementary Figure S2
Download PDF (130.8 KB)Acknowledgments
We thank Drs Sheli Radoshitzky (Molecular and Translational Sciences, USAMRIID) and Jens Kuhn (NIH/NIAID Integrated Research Facility at Fort Detrick (IRF-Frederick), MD; USA) for their careful manuscript review. We also thank Dr Andres Salazar (Scientific Director) of Oncovir (Washington, DC, USA) for providing the clinical-grade dsRNA adjuvant poly-ICLC (Hiltonol). This research was conducted under an Institutional Animal Care and Use Committee-approved protocol in compliance with the US Animal Welfare Act, Public Health Service Policy, and other federal statutes and regulations relating to animals and experiments involving animals. The facility in which this research was conducted (USAMRIID) is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and adheres to the principles stated in the Guide for the Care and Use of Laboratory Animals, by the National Research Council, 2011. This research was funded by grants provided by the US Department of Defense’s Defense Threat Reduction Agency (DTRA).
Supplementary Information for this article can be found on the Emerging Microbes & Infections website (http://www.nature.com/emi)
Related Research Data
References
- Wong G, Kobinger GP.Backs against the wall: novel and existing strategies used during the 2014-2015 Ebola virus outbreak. Clin Microbiol Rev 2015;28: 593–601.
- Marzi A, Feldmann H.Ebola virus vaccines: an overview of current approaches. Expert Rev Vaccines. 2014;13: 521–531.
- Cooper CL, Bavari S.A race for an Ebola vaccine: promises and obstacles. Trends Microbiol 2015;23: 65–66.
- De Santis O, Audran R, Pothin Eet al.Safety and immunogenicity of a chimpanzee adenovirus-vectored Ebola vaccine in healthy adults: a randomised, double-blind, placebo-controlled, dose-finding, phase 1/2a study. Lancet Infect Dis 2016;16: 311–320.
- Huttner A, Dayer JA, Yerly Set al.The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: a randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect Dis 2015;15: 1156–1166.
- Regules JA, Beigel JH, Paolino KMet al.A recombinant vesicular stomatitis virus ebola vaccine—preliminary report. N Engl J Med 2017;376: 330–341.
- Martins KA, Jahrling PB, Bavari Set al.Ebola virus disease candidate vaccines under evaluation in clinical trials. Expert Rev Vaccines 2016;15: 1101–1112.
- Falzarano D, Geisbert TW, Feldmann H.Progress in filovirus vaccine development: evaluating the potential for clinical use. Expert Rev Vaccines 2011;10: 63–77.
- Krause PR, Bryant PR, Clark Tet al.Immunology of protection from Ebola virus infection. Sci Transl Med 2015;7: 286ps11.
- Sullivan NJ, Martin JE, Graham BSet al.Correlates of protective immunity for Ebola vaccines: implications for regulatory approval by the animal rule. Nat Rev Microbiol 2009;7: 393–400.
- Bradfute SB, Bavari S.Correlates of immunity to filovirus infection. Viruses 2011;3: 982–1000.
- Wong G, Kobinger GP, Qiu X.Characterization of host immune responses in Ebola virus infections. Expert Rev Clin Immunol 2014;10: 781–790.
- Sullivan NJ, Hensley L, Asiedu Cet al.CD8+ cellular immunity mediates rAd5 vaccine protection against Ebola virus infection of nonhuman primates. Nat Med 2011;17: 1128–1131.
- Olinger GG, Bailey MA, Dye JMet al.Protective cytotoxic T-cell responses induced by venezuelan equine encephalitis virus replicons expressing Ebola virus proteins. J Virol. 2005;79: 14189–14196.
- Warfield KL, Olinger GG.Protective role of cytotoxic T lymphocytes in filovirus hemorrhagic fever. J Biomed Biotechnol 2011;2011: 984241.
- Wong G, Richardson JS, Pillet Set al.Immune parameters correlate with protection against ebola virus infection in rodents and nonhuman primates. Sci Transl Med 2012;4: 158ra46.
- Marzi A, Engelmann F, Feldmann Fet al.Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc Natl Acad Sci USA 2013;110: 1893–1898.
- Jones SM, Stroher U, Fernando Let al.Assessment of a vesicular stomatitis virus-based vaccine by use of the mouse model of Ebola virus hemorrhagic fever. J Infect Dis 2007;196: S404–S412.
- Martins KA, Cooper CL, Stronsky SMet al.Adjuvant-enhanced CD4 T cell responses are critical to durable vaccine immunity. EBioMedicine 2016;3: 67–78.
- Martins KA, Steffens JT, van Tongeren SAet al.Toll-like receptor agonist augments virus-like particle-mediated protection from Ebola virus with transient immune activation. PLoS ONE 2014;9: e89735.
- Warfield KL, Olinger G, Deal EMet al.Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J Immunol 2005;175: 1184–1191.
- Blaney JE, Marzi A, Willet Met al.Antibody quality and protection from lethal Ebola virus challenge in nonhuman primates immunized with rabies virus based bivalent vaccine. PLoS Pathog 2013;9: e1003389.
- Qiu X, Audet J, Wong Get al.Sustained protection against Ebola virus infection following treatment of infected nonhuman primates with ZMAb. Sci Rep 2013;3: 3365.
- Qiu X, Wong G, Audet Jet al.Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014;514: 47–53.
- Pettitt J, Zeitlin L, Kim DHet al.Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci Transl Med 2013;5: 199ra13.
- McHeyzer-Williams M, Okitsu S, Wang Net al.Molecular programming of B cell memory. Nat Rev Immunol 2012;12: 24–34.
- Mesin L, Ersching J, Victora GD.Germinal center B cell dynamics. Immunity 2016;45: 471–482.
- Bengtsson KL, Song H, Stertman Let al.Matrix-M adjuvant enhances antibody, cellular and protective immune responses of a Zaire Ebola/Makona virus glycoprotein (GP) nanoparticle vaccine in mice. Vaccine 2016;34: 1927–1935.
- Warfield KL, Dye JM, Wells JBet al.Homologous and heterologous protection of nonhuman primates by Ebola and Sudan virus-like particles. PLoS ONE 2015;10: e0118881.
- Warfield KL, Bosio CM, Welcher BCet al.Ebola virus-like particles protect from lethal Ebola virus infection. Proc Natl Acad Sci USA 2003;100: 15889–15894.
- Warfield KL, Swenson DL, Olinger GGet al.Ebola virus-like particle-based vaccine protects nonhuman primates against lethal Ebola virus challenge. J Infect Dis 2007;196: S430–S437.
- Cazares LH, Ward MD, Brueggemann EEet al.Development of a liquid chromatography high resolution mass spectrometry method for the quantitation of viral envelope glycoprotein in Ebola virus-like particle vaccine preparations. Clin Proteomics 2016;13: 18.
- De Silva NS, Klein U.Dynamics of B cells in germinal centres. Nat Rev Immunol 2015;15: 137–148.
- Ma CS, Deenick EK, Batten Met al.The origins, function, and regulation of T follicular helper cells. J Exp Med 2012;209: 1241–1253.
- Baumjohann D, Preite S, Reboldi Aet al.Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 2013;38: 596–605.
- Elgueta R, Benson MJ, de Vries VCet al.Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 2009;229: 152–172.
- Kawabe T, Naka T, Yoshida Ket al.The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1994;1: 167–178.
- Muramatsu M, Kinoshita K, Fagarasan Set al.Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000;102: 553–563.
- Kitamura D, Roes J, Kuhn Ret al.B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 1991;350: 423–426.
- Allio T.Product development under FDA's animal rule: understanding FDA's expectations and potential implications for traditional development programs. Therap Innov Regul Sci 2016;50: 660–670.
- Bray M, Davis K, Geisbert Tet al.A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis 1998;178: 651–661.
- Nakayama E, Saijo M.Animal models for Ebola and Marburg virus infections. Front Microbiol 2013;4: 267.
- Bradfute SB, Anthony SM, Stuthman KSet al.Mechanisms of immunity in post-exposure vaccination against Ebola virus infection. PLoS ONE 2015;10: e0118434.
- Bradfute SB, Warfield KL, Bray M.Mouse models for filovirus infections. Viruses 2012;4: 1477–1508.
- Furuyama W, Marzi A, Nanbo Aet al.Discovery of an antibody for pan-ebolavirus therapy. Sci Rep 2016;6: 20514.
- Warfield KL, Perkins JG, Swenson DLet al.Role of natural killer cells in innate protection against lethal ebola virus infection. J Exp Med 2004;200: 169–179.
- Nanton MR, Way SS, Shlomchik MJet al.Cutting edge: B cells are essential for protective immunity against Salmonella independent of antibody secretion. J Immunol 2012;189: 5503–5507.
- Misumi I, Whitmire JK.B cell depletion curtails CD4+ T cell memory and reduces protection against disseminating virus infection. J Immunol 2014;192: 1597–1608.