
Abstract
The recent convergence of genetic elements encoding phenotypic carbapenem-resistance and hypervirulence within a single Klebsiella pneumoniae host strain represents a major public concern. To obtain a better understanding of the genetic characteristic of this emerging ‘superbug’, the complete genomes of 3 isolates of ST11 carbapenemase-producing hypervirulent K. pneumoniae were generated using the Oxford nanopore MinION platform. Comparative whole-genome analysis identified 13 SNPs and 3 major regions of indels in the chromosomes of the clonally disseminated isolates. ISKpn18-mediated disruption in the mgrB gene, which was associated with colistin resistance, was identified in two later strains, leading to the emergence of hypervirulent K. pneumoniae that was simultaneously colistin- and carbapenem-resistant. Five plasmids were recovered from each isolate, including a 178 Kb IncHI1B/FIB-type rmpA2-bearing virulence plasmid, a 177.5 Kb IncFII/R self-transferable blaKPC-2-bearing MDR plasmid, a 99.7 Kb Incl1 plasmid and two ColRNAI-type plasmids of sizes of 11.9 and 5.6 Kb, respectively. The presence of homologous regions between the non-conjugative virulence plasmid and conjugative blaKPC-2-bearing MDR plasmid suggests that transmission of the virulence plasmid from ST23 K. pneumoniae to ST11 CRKP may be mediated by the co-integrated transfer of these two plasmids. Emergence of colistin-resistant and carbapenemase-producing hypervirulent K. pneumoniae strains further emphasizes the urgency for the establishment of a coordinated global program to eradicate hypervirulent and/or pan-drug-resistant strains of K. pneumoniae from clinical settings and the community.
Introduction
Hypervirulent Klebsiella pneumoniae (hvKP), primarily associated with sequence type (ST) 23, and carbapenem-resistant (CR) K. pneumoniae, mostly belonging to ST11, represent two major types of clinically significant pathogens in ChinaCitation1,Citation2. Recently, a genetically and phenotypically convergent clone that simultaneously exhibits hypervirulence and carbapenem resistance, namely CR-hvKP, has emerged. Apart from the ST 23 and ST11 strains, CR-hvKP strains of less common genetic types such as ST65, ST1797, ST43, and ST231, have also been identifiedCitation3–Citation8. These strains are considered real ‘superbugs’ as they are not only hypervirulent and multidrug resistant, but also highly transmissible, causing severe and often fatal infections in both hospital settings and the communityCitation6. According to the announced draft genomes of representative CR-hvKP isolates, such convergent clones form as a result of either horizontal transfer of resistance plasmids to hypervirulent strains or through acquisition of the pLVPK-like virulence plasmid by carbapenemase-producing strainsCitation3,Citation6. In a previous study, we reported a fatal outbreak of ST11 CR-hvKP strains in a Chinese hospital and revealed the genomic characteristics of five representative causative strains based on Illumina short read sequencesCitation6. The strains were demonstrated to belong to one single clone with slightly different PFGE patterns (one or two band differences) among the clone strains, suggesting the possibility that genomic rearrangement frequently occurs among these strainsCitation6. However, the complete genome sequence of the CR-hvKP strain is not currently available, preventing comprehensive and detailed genetic analysis of this ‘superbug’.
Recent advances in third generation sequencing platforms, including the single molecule real-time (SMRT) sequencing (Pacific Biosciences), nanopore sequencing (Oxford Nanopore Technologies), etc., have provided effective tools for delineating the complete sequences of bacterial genomesCitation9. In particular, nanopore MinION sequencing offers the advantage of being a timesaving procedure for library preparation and reliable data analysisCitation10. Through hybrid assembly with short-read sequencing data and MinION nanopore reads, high-quality and completely assembled sequences can be generatedCitation10. To track microevolution events among the outbreak strains as well as provide reference sequences for genome-based hvKP studies, we performed long read sequencing with the portable Oxford Nanopore MinION device and delineated the complete genetic structures of these ST11 CR-HvKP ‘superbugs’.
Results
Between late February and April 2016, five CR K. pneumoniae strains that caused fatal infections in five patients were identified at the integrated ICU of the Second Affiliated Hospital of Zhejiang University (Hangzhou, China)Citation6. Phenotypic analysis showed that the isolates were string test-positive, CR and hypervirulent. Analysis of short-read sequencing data suggested that the isolates originated from a single clone, which belonged to ST11, serotype K47 K. pneumoniae, and harbored the blaKPC-2 gene and a pLVPK-like virulence plasmidCitation6. ST11, being the dominant clone of KPC-producing K. pneumoniae in China, comprises at least three clusters distinguishable mainly by the serotypesCitation2,Citation11. Through acquiring the pLVPK-like virulence plasmid, the ST11 CR-hvKP clone emerged as a real ‘superbug’, that is simultaneously hypervirulent, multidrug resistant, and highly transmissibleCitation3,Citation6. Retrospective studies have demonstrated that CR-hvKP actually emerged sometime before 2015 and has since become detectable in different regions of Asia, including Mainland China, Hong Kong and India, indicating that hvKP may undergo worldwide dissemination in the near futureCitation5,Citation12,Citation13.
General characteristics of the ST11 CR-hvKP genomes
PFGE analysis in a previous study revealed that K. pneumoniae isolates 2, 3, and 5 are more closely related and exhibit almost identical PFGE patterns when compared with isolates 1 and 4Citation6, thus only one of the three isolates (K. pneumoniae 5) as well as isolates 1 and 4 were subjected to long-read sequencing. The complete genome of each of the three isolates was found to contain a chromosome of 5.4 Mbp in size, and 5 plasmids ranging from 5 to 178 Kbp. The overall G + C content of the three chromosomes was 57.4%, with ~5200 coding sequences (CDSs) in each isolate (Supplementary table S1). The three CR-hvKP strains, despite being members of the same clone, were found to harbor different numbers of insertion sequences (ISs) and prophages. On the other hand, the antimicrobial resistance (AMR) genes aadA2, sul1, blaSHV-11 were detectable in all three of the chromosomes, of which blaSHV was a core chromosomal gene in K. pneumoniae, and sul1 and aadA2 were integrated into the chromosome via insertion sequence (IS26)-mediated transposition. The chromosomes of the test strains were also found to harbor the previously described yersinabactin system, the type 1 fimbriae cluster fimABCDE, and the type 3 fimbriae cluster mrkABCD, which encode a moderate level of virulence in K. pneumoniae.
Comparison of the ST11 CR-hvKP chromosomal sequences
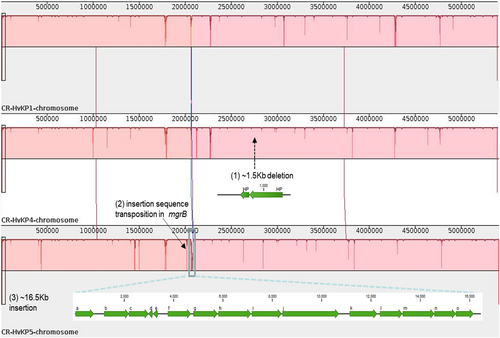
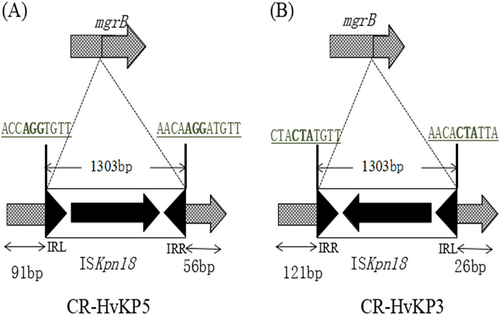
To investigate the population structure of the ST11 isolates in detail, we studied the chromosomal SNPs among the three fully sequenced isolates. By aligning the raw Illumina sequencing reads of isolates 1, 4, and 5 against the complete sequence of K. pneumoniae isolate 1, a total of 13 synonymous and nonsynonymous SNPs were identified (Table ). In addition to SNPs, the ST11 CR-hvKP chromosomes mainly differ from each other in three regions of deletion and insertions (Fig. ), one of which is a 16.5-Kb genomic region that contains 15 predicted ORFs at position 2.06 Mb in the K. pneumoniae 5 genome. A BLASTN search revealed that this element shares 100% identity with several fragments in various other K. pneumoniae genomes with 100% coverage, including the genomes of the ST11 strains SWU01 and GD4Citation11,Citation14, suggesting that this element is widely distributed, but is not necessarily a core cassette in K. pneumoniae genomes. Genes related to threonine and serine metabolism and transport during anaerobic growth (the tdc operon) and a cluster encoding branched-chain amino acid ABC transporter (the liv operon) were annotated in this 16.5-Kb elementCitation15,Citation16. Additionally, a 1.5-Kb region found at position 2.7 Mb of K. pneumoniae 1 and 5 was absent in the genome of K. pneumoniae 4. This element was predicted to encode two hypothetical proteins. Furthermore, truncation of the mgrB gene by ISKpn18 at position 2.0 Mb was detected in the genome of K. pneumoniae 5. To verify the effect of mgrB gene truncation, colistin susceptibilities of the five isolates were tested. Strains K. pneumoniae 3 and 5 were both colistin-resistant, with MICs of 64 µg/ml. Genetic analysis revealed that the mgrB genes in the two strains were both truncated by ISKpn18, but the positions (nt91 and nt121) and orientations of the two insertions were different (Fig. ). To track the origin of this IS element, which frequently causes colistin resistance in K. pneumoniae, we searched the chromosome of these three strains for the presence of this IS element. Our results indicated that the ISKpn18 element was located at position 4.4 Mb of the K. pneumoniae chromosome in these three strains.
List of chromosomal SNP differences among the clonal CR-hvKP outbreak strains tested
Major regions of divergence are labeled, including: (1) An 1.5-Kb deletion in the K. pneumoniae 4 genome with two genes encoding hypothetical proteins (indicated with a black arrow); (2) disruption of the mgrB gene by insertion sequence (ISKpn18) in the K. pneumoniae 5 genome (indicated with a black arrow); (3) A 16.5-Kb region of the K. pneumoniae 5 genome (indicated by a blue rectangle) is absent in the other two ST11 K. pneumoniae genomes. Predicted genes within this region are indicated: (a) peptidase; (b) fatty acid desaturase; (c) phosphate ABC transporter substrate-binding protein; (d) nitrilotriacetate monooxygenase; (e) hypothetical protein; (f) transcriptional regulator TdcA; (g) bifunctional threonine ammonia-lyase/L-serine ammonia-lyase TdcB; (h) threonine/serine transporter TdcC; (i) propionate kinase; (j) formate C-acetyltransferase; (k) branched-chain amino acid ABC transporter substrate-binding protein; (l) branched-chain amino acid ABC transporter permease, LivH; (m) branched-chain amino acid ABC transporter permease, LivM; (n) ABC transporter ATP-binding protein, LivG; (o) ABC transporter ATP-binding protein, LivF
The mgrB gene was truncated by ISKpn18 in both K. pneumoniae 5 (a) and K. pneumoniae 3 (b) strains with opposite directions at different positions. Target site duplications are underlined with bold letters. The left and right inverted repeats (IRL and IRR) of ISKpn18 are represented as black triangles
Extrachromosomal elements of the ST11 CR-hvKP isolates
Each of the three ST11 CR-hvKP isolates was found to harbor five plasmids, the sizes of which were 178, 177.5, 99.7, 11.9, and 5.6 Kb, respectively (Fig. , Supplementary table S2). Due to the high degree of sequence similarity of the plasmid contents in the three hvKP isolates, plasmids in strain K. pneumoniae 4 were chosen as representative sequences in subsequent analyses. The pLVPK-like virulence plasmid (pVir-CR-HvKP, 178 Kb) was only slightly larger than another blaKPC-2-bearing MDR plasmid in the same strain (pKPC-CR-HvKP, 177.5 Kb). A BLASTN search in the NCBI database revealed that pVir-CR-HvKP4 exhibited 99% identity to the 219 Kb typical virulence plasmid in K. pneumoniae, pLVPK (AY378100), with 90% coverage. As reported previously, a 58.8-Kb element that encodes hypothetical proteins located between two genes in position 75.6 Kb of pLVPK was absent in pVir-CR-HvKP4 (Supplementary Figure S1)Citation6. This element carries 71 predicted ORFs, including rmpA, iroBCDN (salmochelin), fecIRA-like ferric citrate uptake-related genes, and 40 other genes with unknown functions. Other major differences between the two plasmids include the 11.8-Kb and 6.7-Kb fragments carried by pVir-CR-HvKP4 but not by pLVPK. This 11.8-Kb fragment, located between the cus locus and a hypothetical gene, shares 100% identity and 100% coverage with a fragment in the 231 Kb K. pneumoniae virulence plasmid, pSGH10Citation17. It carries 20 ORFs, including multiple insertion sequences (an IS1R, a truncated ISEc27 and 2 IS102-like sequences) and genes encoding hypothetical proteins. Likewise, the 6.7-Kb element carries an IS5075, two IS102-like insertion sequences and genes of unknown functions. The 6.7-Kb element, along with a downstream 4.7-Kb fragment of pSGH10, exhibited 99% identity and 99% coverage with an 11.4-Kb fragment in plasmid pKPC-CR-HvKP4 (Fig. ). Additionally, inversion at a 27.6-Kb fragment carrying multiple heavy metal resistance genes (the sil, pco and pbr gene clusters) was detected in plasmid pVir-CR-HvKP4 when compared with pLVPK. The pbr locus was located in a Tn3-like transposon, which could potentially contribute to the genetic rearrangement of these virulence plasmids.
Each of the three ST11 isolates carried five plasmids, with sizes of 178, 177.5, 99.7, 11.9, and 5.6 Kb. Insertion sequences and antimicrobial resistance genes are annotated with blue and yellow fonts, respectively. Plasmid name, size, and replicon types are depicted in the middle of each circle. The 11.4-Kb homologous region (HR) in the virulence plasmid and MDR plasmid, and 4.5-Kb region of divergence (RD) in the 99.7-Kb plasmid are highlighted
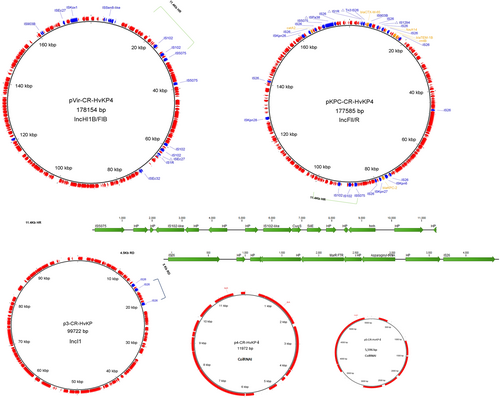
The 177.5-Kb blaKPC-2-bearing plasmid was found to belong to the class of IncFII/R-type self-transferable MDR plasmid and carry the resistance genes blaKPC-2, blaCTX-M-65, blaTEM-1b, rmtB, catA2 and fosA14. This plasmid shares 94% coverage and 99% identity to the K. pneumoniae IncFII/R type plasmid pKPGD4 (GenBank accession: CP025952), which was previously isolated from an ST11 isolate and also found to harbor the aforementioned resistance genesCitation11. The major difference between plasmids pKPC-CR-HvKP4 and pKPGD4 lies in the 11.4-Kb element, which is carried by the plasmids pKPC-CR-HvKP4 and pVir-CR-HvKP4, but absent in pKPGD4 (Fig. , supplementary Figure S2).
The 99.7-Kb Incl1-type, non-MDR plasmid p3-CR-HvKP4 exhibits 94% coverage and 99% identity to the 115.7-Kb E. coli Incl1-type plasmid pS68 (KU130396). Plasmid p3-CR-HvKP4 shares 94 Kb of its sequence with pS68. However, a 22.4-Kb region of pSa68 bordered by IS26 at both ends, which carries multiple drug resistance genes (aadA1, rmtE, ermB), is lost in the 99.7-Kb plasmid. Instead, a 4.5-Kb region that includes 9 ORFs was integrated into the pS68 backbone. This region encodes a MarR family transcriptional regulator, an asparaginyl-tRNA synthetase, two IS26 transposases and 5 hypothetical proteins. It is 100% identical to the 277-Kb plasmid pRpNDM1-1 (JX515588), which was previously isolated from a Raoultella planticola isolateCitation18.
Furthermore, all ST11 CR-hvKP isolates were found to carry two ColRNAI plasmids of sizes 11.9 and 5.6-Kb. The 11.9-Kb plasmid was found to exhibit 99% identity to an unnamed plasmid (CP023932) isolated from K. pneumoniae strain FDAARGOS_443 with 100% coverage, and harbor 13 ORFs encoding colicin E3, a mobilization protein and hypothetical proteins. The 5.6-Kb plasmid was commonly detected in K. pneumoniae, and encodes the RelE/StbE replicon stabilization toxin/antitoxin system and seven hypothetical proteins.
Discussion
The K. pneumoniae genome is known to undergo constant evolution, with an estimated 10.1 nucleotide substitutions/genome/yearCitation19,Citation20. Despite being members of a single clone that were indistinguishable by phylogenetic analysis and simple sequence alignments (Fig. ), the outbreak-related ST11 isolates differ by 13 specific nucleotides termed SNPs. Given that the three isolates were sampled within a 2-month-period, we estimated that the ST11 clone could have evolved at a velocity higher than that reported previouslyCitation20. Apart from SNPs, mobile genome elements including plasmids, phages, integrated conjugative elements (ICEs) and insertion sequences (ISs), which have been demonstrated to drive genomic evolution in K. pneumoniae, were not identical in all three isolates, indicating that active genomic rearrangement occurred during the transmission processCitation21. In line with this finding is that three major indels were detected among the three isolates, including genes related to amino acid metabolism and transportation, hypothetical genes and truncation of the mgrB gene by ISKpn18. MgrB is a negative regulator of the PhoPQ two-component regulatory system, which is associated with colistin resistance in K. pneumoniaeCitation22. Inactivation of the mgrB gene was demonstrated to be a common cause of colistin resistance in clinical carbapenemase-producing K. pneumoniae strainsCitation23. A previous study reported that colistin resistance in a ST258 blaKPC-2-bearing K. pneumoniae strain was mediated by insertion of ISKpn18 in the mgrB geneCitation24. Also, insertion of ISKpn18 in the ramR gene, which is associated with tigecycline resistance in K. pneumoniae, has been reportedCitation25. A previous study has demonstrated that ISKpn18 is almost entirely restricted to strains of CG258 and the proportion of intragenic insertion of ISKpn18 is over 80% in K. pneumoniaeCitation26. All evidence indicates that ISKpn18 can readily cause intragenic insertion in K. pneumoniae. Because of this inactivation, two later strains, K. pneumoniae 3 and 5, have emerged as real ‘superbugs’; they are simultaneously carbapenem- and colistin- resistant, hypervirulent and transmissible and could not be treated with last-line-of-defense antibiotics.
Plasmids play pivotal roles in the dissemination of virulence determinants and antimicrobial resistance phenotypes. In this study, five plasmids with sizes of 178, 177.5, 99.7, 11.9, and 5.6 Kb were identified in each of the three sequenced genomes. The 178-Kb plasmid pVir-CR-HvKP4 is a pLVPK-like virulence plasmid. pLVPK is known to harbor genes encoding capsular polysaccharide synthesis regulators (rmpA and rmpA2) and iron-acquisition systems (iucABCDiutA and iroBCDN siderophore gene clusters), which are associated with enhanced virulence potential and genes related to heavy metal (copper, silver, lead, and tellurite) resistanceCitation27. Despite the absence of a 58.8-Kb element carrying multiple virulence-related genes from pVir-CR-HvKP4 compared with plasmid pLVPK, the plasmid does contribute to the virulence of the host strain. Curing of plasmid pVir-CR-HvKP4 resulted in a negative string test phenotype and substantially reduced virulence potential of the host strainCitation6. Multiple mobile elements were detected on pVir-CR-HvKP4 and genetic rearrangements were found compared with other related virulence plasmids, which suggests that this virulence plasmid has undergone constant evolution by acquisition and loss of DNA fragments through horizontal gene transfer mediated by different IS elements. Future research should study whether variation in the genetic organization of virulence plasmids leads to the divergence in the virulence potential of K. pneumoniae isolates. The 177.5-Kb blaKPC-2-bearing conjugative plasmid pKPC-CR-HvKP4 contributed to the carbapenem resistance phenotypes of the host strainsCitation6. Plasmids that possess the pKPC-CR-HvKP4 backbone are highly prevalent among blaKPC-2-bearing K. pneumoniae strains in China according to previous studiesCitation11. A 11.4-Kb element on pKPC-CR-HvKP4 was homologous to that on pVir-CR-HvKP4, suggesting that it might contribute to the transmission of a virulence plasmid that is not conjugative from ST23 HvKP to ST11 K. pneumoniae with the help of the conjugative blaKPC-2-bearing plasmid, likely through plasmid cointegration. The details of the mechanisms of transmission of virulence plasmids require further investigation.
In summary, this study delineated genetic microevolution events that occurred among clonal ST11 hypervirulent, CR K. pneumoniae strains by using the nanopore MinION sequencing platform. We identified various chromosomal SNPs, indels and unique genetic features of the plasmids harbored by the tested strains. To our knowledge, this is not only the first report of the complete sequence of a ST11 CR-hvKP clone but also the first report on the emergence of colistin- and CR hypervirulent K. pneumoniae strains. In addition, through sequence analysis of their extrachromosomal plasmids, the mechanisms of transmission of virulence plasmids from ST23 HvKP to ST11 CRKP might be revealed. Coordinated efforts are required to eradicate such hypervirulent and pan-drug-resistant strains, which may otherwise pose a worldwide public health threat in the near future.
Material and Methods
Strains and susceptibility tests
Source information of the five outbreak strains studied in this work can be found in our previous studyCitation6. The colistin susceptibility of the isolates was determined and interpreted as previously describedCitation28.
Whole genome sequencing and assembly
Three of the five outbreak-related K. pneumoniae isolates (1, 4, and 5) which displayed different PFGE patterns as described in our previous study were selected for whole genome sequencing using the nanopore MinION device (Oxford Nanopore Technologies, Oxford, United Kingdom), according to previously described methodsCitation10. Briefly, genomic DNA was extracted from overnight cultures using the PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA, USA). MinION libraries of the test isolates were prepared using the SQK-RBK001 nanopore sequencing kit (version R9.4) according to the manufacturer’s instructions. Illumina sequencing reads for the isolates were derived from the previous studyCitation6. The hybrid read set (both Illumina and nanopore reads) for the genomes of each isolate was assembled using Unicycler (v0.4.0) with manual curation as necessaryCitation29. The complete genome sequences were annotated with the RAST toolCitation30 and ProkkaCitation31. The chromosome was adjusted, with dnaA being the first gene.
Bioinformatics analysis
Sequences and annotations were visualized and edited with the CLC Genomics Workbench (version 9.0). ICEs, acquired antibiotic resistance genes, plasmid replicons, ISs, virulence-related genes and phage-associated regions were identified using previously described methodsCitation11. Single-nucleotide polymorphisms (SNPs) were detected using Snippy v3.2 (https://github.com/tseemann/snippy) by mapping the Illumina sequence reads of the three CR-hvKP isolates to the complete chromosome sequence of isolate K. pneumoniae 1. A minimum coverage depth of 10 and base call stringency of 90% were selected for SNP detection. The identified SNPs were manually inspected and edited. ProgressiveMauve (version 2.4.0) was applied to compare the chromosomal architecturesCitation32. BLASTN was conducted to screen for sequences homologous to the sequenced plasmids in the NCBI database. Comparison between homologous plasmids was conducted using EasyFig 2.1Citation33. Plasmid maps were generated using the BLAST Ring Image Generator (BRIG) v0.95Citation34.
Data availability
All sequencing data have been deposited in GenBank under the accession numbers SAMN09487516, SAMN09487517, and SAMN09487518.
Supplementary Information
Download PDF (238.1 KB)Acknowledgements
We gratefully acknowledge the financial support of this project by the Collaborative Research Fund of Hong Kong Research Grant Council (C5026-16G).
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41426-018-0146-6).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Research Data
References
- StruveCMapping the Evolution of Hypervirulent Klebsiella pneumoniaemBio20156 e0063010.1128/mBio.00630-154513082
- QiYST11, the dominant clone of KPC-producing Klebsiella pneumoniae in ChinaJ. Antimicrob. Chemother.201166307 31210.1093/jac/dkq431
- Chen, L. & Kreiswirth B. N. Convergence of carbapenem-resistance and hypervirulence in Klebsiella pneumoniae. Lancet Infect. Dis., 18, 2–3 (2018).
- ZhangREmergence of carbapenem-resistant serotype K1 hypervirulent Klebsiella pneumoniae strains in ChinaAntimicrob. Agents Chemother.20156070971110.1128/AAC.02173-154704206
- Shankar, C. et al. Draft genome sequences of three hypervirulent carbapenem-resistant Klebsiella pneumoniae isolates from bacteremia. Genome Announc., 4, e01081–16 (2016).
- Gu, D. et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect. Dis., 2017.
- YaoBClinical and molecular characteristics of multi-clone carbapenem-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in a tertiary hospital in Beijing, ChinaInt. J. Infect. Dis.20153710711210.1016/j.ijid.2015.06.023
- ZhangYEmergence of a hypervirulent carbapenem-resistant Klebsiella pneumoniae isolate from clinical infections in ChinaJ. Infect.20157155356010.1016/j.jinf.2015.07.010
- GoodwinSMcPhersonJDMcCombieWRComing of age: ten years of next-generation sequencing technologiesNat. Rev. Genet.20161733310.1038/nrg.2016.49
- LiREfficient generation of complete sequences of MDR-encoding plasmids by rapid assembly of MinION barcoding sequencing dataGigascience2018719
- Dong, N. et al. Genome analysis of Clinical Multilocus Sequence Type 11 Klebsiella pneumoniae from China. Microb. Genom., 10.1099/mgen.0.000149 (2018).
- WongMHYEmergence of carbapenem-resistant hypervirulent Klebsiella pneumoniaeLancet Infect. Dis.2018182410.1016/S1473-3099(17)30629-1
- DuPZhangYChenCEmergence of carbapenem-resistant hypervirulent Klebsiella pneumoniaeLancet Infect. Dis.201818232410.1016/S1473-3099(17)30625-4
- ZhangLWhole-genome sequence of a carbapenem-resistant hypermucoviscous Klebsiella pneumoniae isolate SWU01 with capsular serotype K47 belonging to ST11 from a patient in ChinaJ. Glob. Antimicrob. Resist.201711878910.1016/j.jgar.2017.09.001
- LaiYCPengHLChangHYIdentification of genes induced in vivo during Klebsiella pneumoniae CG43 InfectionInfect. Immun.2001697140714510.1128/IAI.69.11.7140-7145.2001100105
- BronnerDClarkeBRWhitfieldCIdentification of an ATP‐binding cassette transport system required for translocation of lipopolysaccharide O‐antigen side‐chains across the cytoplasmic membrane of Klebsiella pneumoniae serotype O1Mol. Microbiol.19941450551910.1111/j.1365-2958.1994.tb02185.x
- Lam, M. M. et al. Population genomics of hypervirulent Klebsiella pneumoniae clonal group 23 reveals early emergence and rapid global dissemination. bioRxiv, 10.1101/225359 (2017).
- LiJSequential isolation in a patient of Raoultella planticola and Escherichia coli bearing a novel ISCR1 element carrying blaNDM-1PLoS One20149e8989310.1371/journal.pone.00898933940617
- MathersAJKlebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae at a single institution: insights into endemicity from whole-genome sequencingAntimicrob. Agents Chemother.2015591656166310.1128/AAC.04292-144325807
- ZautnerAEMonitoring microevolution of OXA-48-producing Klebsiella pneumoniae ST147 in a hospital setting by SMRT sequencingJ. Antimicrob. Chemother.2017722737274410.1093/jac/dkx216
- ChenLCarbapenemase-producing Klebsiella pneumoniae: molecular and genetic decodingTrends Microbiol.20142268669610.1016/j.tim.2014.09.0034365952
- PoirelLThe mgrB gene as a key target for acquired resistance to colistin in Klebsiella pneumoniaeJ. Antimicrob. Chemother.201470758010.1093/jac/dku323
- CannatelliAMgrB inactivation is a common mechanism of colistin resistance in KPC carbapenemase-producing Klebsiella pneumoniae of clinical originAntimicrob. Agents Chemother.2014585696570310.1128/AAC.03110-144187966
- ArenaFColistin resistance caused by inactivation of the MgrB regulator is not associated with decreased virulence of sequence type 258 KPC carbapenemase-producing Klebsiella pneumoniaeAntimicrob. Agents Chemother.2016602509251210.1128/AAC.02981-154808163
- HudsonCMResistance determinants and mobile genetic elements of an NDM-1-encoding Klebsiella pneumoniae strainPLoS One20149e9920910.1371/journal.pone.00992094048246
- AdamsMDWrightMSBishopBQuantitative assessment of insertion sequence impact on bacterial genome architectureMicrobial Genomics20162e00006210.1099/mgen.0.0000625343135
- ChenYTSequencing and analysis of the large virulence plasmid pLVPK of Klebsiella pneumoniae CG43Gene200433718919810.1016/j.gene.2004.05.008
- LiRComplete genetic analysis of plasmids carrying mcr-1 and other resistance genes in an Escherichia coli isolate of animal originJ. Antimicrob. Chemother.201772696699
- WickRRUnicycler: resolving bacterial genome assemblies from short and long sequencing readsPLoS. Comput. Biol.201713e100559510.1371/journal.pcbi.10055955481147
- OverbeekRThe SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST)Nucleic Acids Res.201442D1D206D21410.1093/nar/gkt1226
- SeemannTProkka: rapid prokaryotic genome annotationBioinformatics2014302068206910.1093/bioinformatics/btu153
- DarlingACMauve: multiple alignment of conserved genomic sequence with rearrangementsGenome Res.2004141394140310.1101/gr.2289704442156
- SullivanMJPettyNKBeatsonSAEasyfig: a genome comparison visualizerBioinformatics2011271009101010.1093/bioinformatics/btr0393065679
- Nabil-Fareed AlikhanNKPNouriLBenZakourBeatsonScottABLAST Ring Image Generator (BRIG): simple prokaryote genome comparisonsBMC Genomics20111210.1186/1471-2164-12-4023163573