
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The present work highlights opportunities and open research issues about the phenomena accompanying the Zr corrosion and describes that with both, the generalization of established opinions and the presentation of individual investigations. The overview aimed to lay out the phenomena accompanying Zr corrosion to better understand the restrictions and assumptions of modern process models and reveal perspectives of simulations. The formation and destruction of the protective oxidized cladding were analyzed and summarized in the following groups: chemical composition of the alloy; kinetics of oxide layer formation; electrochemical processes; chemistry of the coolant; hydrogen absorption; irradiation. The physicochemical, electrochemical, and crystallographic processes driving the multi-scale, multi-phase, multi-parametric Zr corrosion are strongly correlated, barely distinguishable, and have combined, and competing impacts that are very comprehensive to the numerical modeling. Despite a boundless number of studies, mostly empirical, Zr corrosion does not still have fundamental analytical descriptions. The noticeable improvement could be achieved by applying modern machine learning technologies allowing the analysis and optimization of multi-parametrical processes. The described groups of phenomena can be included in the clustering of the neural network of the deep learning approach in the integrated expert system for the Zr alloy corrosion.
1 Introduction
Zirconium alloys are used extensively for the nuclear fuel cladding that forms the fuel rods. Due to their small neutron absorption cross-section, alloys containing more than 97 wt% zirconium, have good resistance to high-temperature corrosion, adequate mechanical properties, and resistance to radiation damage. The reason for the corrosion resistance of the alloy should be attributed to the presence of a thin protected (‘passive’) film of oxide on the alloy surface. The formation, stability (constancy of the layer thickness and structure), and destruction of the passive films are very important issues in metal technology. The passive surfaces are susceptible to various forms of degradation: pitting corrosion, stress corrosion cracking, corrosion fatigue, expansion of the carrier metal, thermal stresses (due to differences in thermal expansion of passive layer and metal), fluid flow, and cavitation (FAC, Flow Accelerated Corrosion), electro- and chemically induced phenomena. The presented work was initiated with an attempt to evaluate the actual possibilities, limitations, and level of idealization of the modern numerical models of the Zr corrosion. We aimed to lay out and highlight opportunities and open research issues about the phenomena accompanying the Zr corrosion and describe that through the generalization of established opinions and a comprehensive vision, and also through the presentation of individual investigations to illustrate the main feature, elucidations, the key factors, and hypotheses, and, sometimes, the conflicts of conclusions from different work groups. Thus, this overview is the preliminary work for the forthcoming analysis of the modern models for Zr corrosion.
Despite intensive examination of different factors influencing the destruction of the protective layer [Citation1–12], an unmanageable number of investigations, and considerable improvements in the fundamental understanding of the multi-parametrical, multi-scale, and multi- phases process of the Zr oxide formation mechanisms, the kinetics of growth and disturbances of the protective layer does not still have fundamental analytical or even semi-analytical description which accounts for at least the most important relationships between the physicochemical, electrochemical, and crystallographic processes driving the oxide-film growth.
Recent progress in large-scale, multi-physics simulation tools, high-resolution experiments, and recent advances in imaging/measuring techniques, sensors, and other important data sources from classical process control and monitoring has led to the generation of large amounts of data, that offer opportunities for extracting new knowledge and gaining more detailed insight applying modern machine learning techniques. This new paradigm for data-intense analyses opens up new possibilities for scientific investigations based on heuristic approaches to data-driven models and physics-constrained learning. The application of machine learning tools supplied with uncertainty quantification applied to such data can lead to remarkable progress in computational simulations of corrosion processes, in an analysis of the collective multidimensional effect of various factors and parameters on the growth, restructuring, and destruction of the Zr alloy oxide layers.
In this treatment, the most important processes involved in the aqueous corrosion of Zr alloys, and their identifying features were systematized and divided into five main clusters, : composition and manufacturing of the alloys; thermodynamic and electrochemical processes; chemistry of aggressive environment; hydrogen absorption; irradiation. The types of corrosion and specific corrosion processes such as pitting, crevice corrosion, stress corrosion, cracking, and corrosion fatigue were not a matter of this work.
Figure 1. Main factors influencing corrosion through their collective effect.
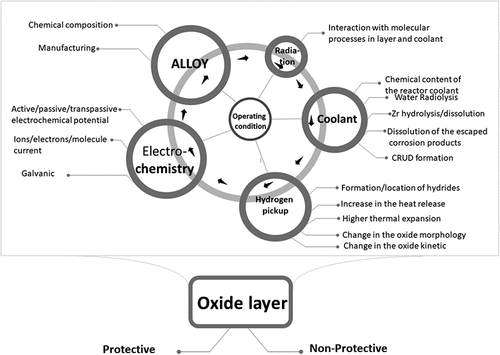
We have included in the manuscript only the published studies, which present the main opinions and trends in the studied area and were very useful for us to highlight and illustrate the process specifications, some effects, or problems. We apologize to the authors and research groups, which were not mentioned – the reason was to reduce the size of the paper. Studies [Citation8, Citation10, Citation13–17] can be recommended for a more in-depth understanding of different related features.
2 Protective oxide layer: properties and formation
In this part, the basic understanding of the zirconium oxide layer formation and growth is briefly presented.
2.1 General physical properties of ZrO2
The volume expansion attendant upon the transformation of Zr→ZrO2 (the Pilling-Bedworth ratio) is 1.56, so the all-covering oxide layer (ZrO2, zirconia, Zirconium dioxide) can be formed. A protective layer is adherent and exhibits a black shiny appearance, whereas a nonprotective oxide is white, nonadherent, and flaky. Besides uniform oxide growth, nodular and shadow corrosion morphologies have been observed [Citation6].
Zirconium oxide exhibits three temperature-dependent polymorphs, namely monoclinic (m-ZrO2), tetragonal (t-ZrO2), and cubic (c-ZrO2) with lattice parameters quite close to each other [Citation18]. The allotropic structure of the ZrO2 lattice is dependent on the temperature, alloying elements, impurities, defects, or doping, all of which are present in the growing oxide. Pressure and grain size have a special impact on the oxide layer structure. As can be seen from , the more compact (higher density) tetragonal zirconia is stabilized with high pressures strongly over m-ZrO2. Therefore, a transition of the tetragonal phase to the monoclinic structure exhibits a volume expansion (i.e. +4–5% of volume strain) which leads to an increase in compression stress in the surrounding matrix [Citation19, Citation20]. The stabilized effect of the grain size is shown in . Below a critical grain size (around 10 nm [Citation21] or between 20 and 30 nm [Citation22]), t-ZrO2 is stabilized over m-ZrO2 whatever the temperature [Citation21, Citation23]. Although the operating temperature of PWRs is significantly lower than the transition temperature for t-ZrO2, the tetragonal phase can be stabilized by stress, sub-stoichiometry, small grain size, pressure, irradiation, and alloying elements.
Figure 2. Reduced stability domains of zirconia allotropic structures depending on temperature and: pressure (left), and grain size (right) [Citation21].
![Figure 2. Reduced stability domains of zirconia allotropic structures depending on temperature and: pressure (left), and grain size (right) [Citation21].](/cms/asset/25dbc88e-0c8a-40c0-b29b-678cbe616bef/tnst_a_2127954_f0002_b.gif)
The zirconium oxide is the n-type semiconductor; therefore, the passive films are characterized by high electrical fields (~106 V/cm) [Citation24]. ZrO2 can demonstrate the properties of an electrolyte: if there is an oxygen concentration difference at the interfaces of the ZrO2 element, a Nernst voltage can be measured.
2.2 Phenomenological kinetics of the zirconia layer formation
Kinetics of the zirconium oxide layer growth, i.e. corrosion rate, is under the impact of several different physical and chemical processes occurring in the bulk metal, on the metal/oxide and oxide/coolant surfaces, and the oxide film. The Zr oxidation and zirconia film growth are driven by the electric field present across the developing oxide layer, and the oxide-film growth is governed by the coupled currents of cations and/or anions, as well as electrons diffusing through the developing oxide film [Citation18, Citation25].
On the bare metal/oxide surface, the redistribution of charges caused by differences between chemical potentials of the metal and oxide layer starts the anodic reactions of Zr oxidation: metal transition to the ionic state
and the O2- (and sometimes OH−) reduction
The anions diffuse through the ZrO2 layer from oxygen vacancies to other ones due to an electrochemical potential gradient. The produced various intermediate non-stoichiometric ZrO2-x oxides reduce the oxygen diffusing through the underlying metallic phases.
On the oxide/coolant interface, the cathodic process of reduction of hydrogen ions or water molecules by diffusing electrons takes place
If dissolved oxygen is present in the solution, in addition to those the oxygen reduction takes place in cathodic processes:
These reactions (2.1)-(2.8) are coupled with energy, heat transfer, adsorption-desorption of molecules from gas/liquid medium and oxide layer, reactions of hydro-peroxide formation or the oxide film dissociation, surface charge, heterogeneous radiolysis in the oxide film pores, the crystallographic and mechanistic transformations of the film oxide, etc. The presence of impurity atoms in the oxide field affects a difference in chemical potentials, types of conduction – electronic or holes, distribution of electric field, and therefore the ion transport through the oxide/metal interface.
The competition between electrochemical and crystallographic-mechanical processes in the oxide layer results in cyclic growth of the oxide film, , an alternation of the protective layer formation and destruction (transition) leading to the multilayer structure of the oxide film. The numbers of repeating cycles (and layers in a film) through the operation/exposition time, and the duration of individual cycles are different for different alloys. The rate of oxidation within a cycle varies. It is assumed that each cycle has two regions: pre-transition and post-transition [Citation26–31].
Figure 3. Short-time corrosion weight gain of Zircaloy-4 in water at 633 K, from [Citation32]. Cycles of kinetics are marked with 30, 31,31.
![Figure 3. Short-time corrosion weight gain of Zircaloy-4 in water at 633 K, from [Citation32]. Cycles of kinetics are marked with 30, 31,31.](/cms/asset/d2cf592f-5d80-40c7-aa05-a8a861f3cb40/tnst_a_2127954_f0003_b.gif)
To highlight the main stages of this process, we modified the illustrations proposed by Wei et al. [Citation33], .
The oxide layer formation starts, when Zr cations under the influence of the Mott potential [Citation34] move from metal towards the metal/oxide interface [Citation26, Citation34–38], with a rapid formation of the intermediate layer, consisting of mainly equiaxed, randomly oriented, monoclinic grains of Zr-enriched oxides, identified as ZrOx [Citation39], with a large number of defects (i.e. oxygen vacancies), .
Figure 4. Schematic presentation of the grain transformation in the pre-transition phase, pore formation, and transition from the first cycle to the second one at the ZrO2 growth. The amount of tetragonal and monoclinic grains is only a schematic, not a quantitative representation. Modified from [Citation33].
![Figure 4. Schematic presentation of the grain transformation in the pre-transition phase, pore formation, and transition from the first cycle to the second one at the ZrO2 growth. The amount of tetragonal and monoclinic grains is only a schematic, not a quantitative representation. Modified from [Citation33].](/cms/asset/1f24a299-606d-44cf-b1c6-e866d7da4c66/tnst_a_2127954_f0004_oc.jpg)
Then this interfacial layer is further transformed into an adjacent sub-layer of stoichiometric ZrO2 composed of m-ZrO2 and t-ZrO2, . As the crystallographic form of the new growing oxide film should be adjusted to the hexagonal lattice of metal [Citation35] that goes through deformation of the monoclinic zirconia m-ZrO2, and arising of the tetragonal zirconia, t-ZrO2, which better correlates with hexagonal lattice and accumulates near the oxidation progression front, the oxide/metal interface, . During continued growth in the pretransition period, the oxidation rate decreases, and the oxide grains become columnar to favor the growth of properly oriented grains to minimize the stress accumulation; a highly oriented tetragonal phase is transformed into the monoclinic phase, which is a well-oriented in-plane of the growth, . The results [Citation26] also indicate that the final grain size of the tetragonal phase is smaller than that of the monoclinic phase. The protective oxides are dense, mostly black, and show wider columnar grains, with smaller grain-to-grain misorientations than the non-protective oxides [Citation37].
Figure 5. Schematic of the inward corrosion of Zr and the expected distribution of monoclinic and tetragonal ZrO2 phase. Modified from [Citation33].
![Figure 5. Schematic of the inward corrosion of Zr and the expected distribution of monoclinic and tetragonal ZrO2 phase. Modified from [Citation33].](/cms/asset/e471209c-c3e7-47e0-8b64-1ee635fab769/tnst_a_2127954_f0005_oc.jpg)
The types of formed grains depend on the operating conditions, the chemistry of the environment, microstructure, chemical composition, and manufacturing history of the Zr alloy.
It has been argued that it is the tetragonal phase that acts as a barrier layer for oxygen diffusion; the thickness (indirectly represented by the tetragonal phase fraction) and the integrity of this tetragonal-rich layer are the dominating contributors to corrosion resistance [Citation6, Citation17, Citation33]. But how it follows from the analyzed literature the higher content and larger grain size of t-ZrO2 increase the mechanical stress in the layer, its porosity, and breakaway and therefore can accelerate protective layer destruction.
The next stage – transition kinetics occurs when oxide films on zirconium alloys reach some ‘critical’ oxide thickness (about 1.5 ~ 3 μm depending on alloys and operating conditions) when the resistance of the oxide suddenly fails, and the corrosion rate abruptly increases, reverting to the value observed at the start of the oxidation [Citation6, Citation10, Citation30, Citation40], . It is not spontaneous – the transition thickness is remarkably reproducible for a given alloy and a given corrosion condition [Citation41]. Yilmazbayhan et al. [Citation41] undertook a detailed study of the oxide structures of four Zr-based alloys (Zircaloy-4, ZIRLO™, ZIRLO is a trademark of Westinghouse Electric Co.1 Zr-2.5%Nb and Zr-2.5%Nb-0.5%Cu) formed in water at 360°C, . It is shown that the rate of repetition is very regular and is a characteristic of the alloy. The results also indicate that there is a cyclic variation of the tetragonal and monoclinic oxide across the oxide thickness with a period of the layer thickness; the final grain size of the tetragonal phase is smaller than that of the monoclinic phase and the monoclinic grain size is smaller in Zircaloy-4 and ZIRLO than in the other two studied alloys.
The destruction of the oxide film is mostly explained by mechanical processes [Citation26–31].
Preuss et al. [Citation42] estimated the highest compressive stresses to be about 1200 MPa near the metal/oxide interface and about zero close to the outer surface in an 80 μm thick oxide layer, formed on ZIRLO. The increasing stress eventually causes lateral cracks/porosities formation from the oxide/metal to the oxide/water interface, thus providing easy access to the water and shortening all diffusion paths in the oxide accelerating corrosion rate, causing further rapid growth of the equiaxed grains and subsequence of columnar grains in the new cycle – post-transition region, . De Gabory et al. [Citation39] observed that the oxide, formed right after the transition has occurred, exhibits a layer of small equiaxed grains with high tetragonal oxide fraction, with similar morphology and structure as the first oxide layer (observed near the oxide/water interface). In this way, the multi-layer structure with a cyclic variation of the equiaxed and columnar grains, composed of the tetragonal and monoclinic phases, across the oxide thickness with a period of the layer thickness [Citation26, Citation43], is formed, .
After several kinetic transitions, at a long enough period of observation, the kinetics would enter a region, with an almost linear oxidation rate. This linear corrosion kinetics has also been referred to as breakaway kinetics leading to catastrophic failure of the oxide [Citation6], i.e. transpassive corrosion, see below.
Figure 6. Transmitted light optical micrographs of oxide layers formed in a) ZIRLO and in b) Zircaloy-4 showing the periodic layers associated with successive oxide transitions formed during corrosion. From [Citation41].
![Figure 6. Transmitted light optical micrographs of oxide layers formed in a) ZIRLO and in b) Zircaloy-4 showing the periodic layers associated with successive oxide transitions formed during corrosion. From [Citation41].](/cms/asset/0b177cb2-be7d-4918-af5a-cd14af9b12a4/tnst_a_2127954_f0006_b.gif)
Although many advances have been made in recent years [Citation2, Citation6, Citation8, Citation10, Citation11, Citation13, Citation31, Citation32, Citation34, Citation41, Citation43–47], a complete understanding of the factors starting the oxide transition leading to breakaway is still lacking. Most of the studies are empirical and concentrated on the microstructural, crystallographic, and mechanical processes in the oxide film. From an ensemble of the observations [Citation6, Citation17, Citation26, Citation29, Citation33, Citation37, Citation48, Citation49] it follows that a possible breakdown mechanism of the passive film could result from accumulated mechanical stress, provoked by the crystallographic form transformations of the large fraction of the t- ZrO2 to monoclinic zirconia, m- ZrO2, associated with a 5% volume expansion.
3 Alloy
The specific role of alloying elements is not unambiguous and depends on the process conditions. Alloying elements, the various impurities, different phases of SPPs, and irregularities in the crystal structure define the microstructure of the developing oxide layer, its cracks and porous formation, and the electric field present across the oxide layer. The electric field drives the migration of oxygen atoms, ions, vacancies, and free electrons through the metal and oxide barrier layer, and consequently reaction rates on the metal/oxide and oxide/environment boundaries. Thereby alloying elements define the corrosion kinetics and, finally, the protective or nonprotective properties of the oxide layer.
Investigations of the chemical compositions and effective technologies of alloys manufacturing have very long history and are extensively described in standard classical books [Citation2, Citation50, Citation51] and in the large amount of classified and unclassified literature [Citation1, Citation3–5, Citation7, Citation8, Citation10, Citation13, Citation14, Citation33, Citation40, Citation42, Citation44, Citation45, Citation47, Citation50–65]. Despite that Zr alloys design and manufacturing remain an active area of research. In the following, the major physicochemical properties of alloys and findings on the influence of the composition of the Zr alloys on the corrosion behavior are summarized in a short form.
It is not worse to mention that pure Zr tends to grow an unstable oxide which is very susceptible to break away, but small additions of alloying elements, such as Sn, Fe, Cr, and Ni cause the oxide formed to be protective and stable [Citation2, Citation6, Citation32, Citation34, Citation66].
3.1 Chemical composition and microstructure
The main targets of alloy design are the microstructure with desired mechanical properties, the reduced hydrogen pick-up behavior, and the high corrosion resistance.
It was early recognized [Citation14] that, in contrast to other alloy systems, the corrosion performance of zirconium alloys worsened as the alloy became purer, with the purest metal exhibiting breakaway corrosion. A further unusual feature of the zirconium alloy system is that a very small proportion of alloying element additions (typically less than 0.5%) is sufficient to effect significant changes in corrosion behavior [Citation2, Citation11]. demonstrates the chemical compositions of modern alloys.
Table 1. Composition percentages of the advanced Zr-based alloys (wt %) [Citation19].
The alloying additions and impurities, solute content, Second-Phase Precipitates/particles (SPP), and thermo-mechanical treatment (degree of cold-work/recrystallization and annealing, rolling, etc.) have a different impact on corrosion rates depending on the environment, such as hydrogenated, oxygenated, and LiOH-containing coolants, temperature, system pressure, radiation, and operating conditions.
Alloying elements with a smaller atomic radius compared to zirconium [Citation45], like Sn or Nb, are intensively used to replace zirconium ions in the oxide lattice to increase the possible coordination of the oxide lattice with the metal lattice and to reduce thereby the mechanical stress.
The alloying elements and impurities of the modern zirconium alloys are either fully dissolved in the alloy matrix or dissolved to a large extent, or almost fully precipitated in small intermetallic SPP. The formation of independent oxides of alloying elements with their crystal lattice will lead to an increase in oxide defectiveness. The exact composition of alloys is a mattering, alloying elements, depending on conditions, can have both positive and negative effects on the general cladding material properties, and especially on its resistance to corrosion phenomena.
In , we collected elements, which are used in alloy manufacturing, and the possible impurities. This table was formed while working with the literature for the preparation of this review. In the first column, ‘+,’ elements, which reduce corrosion, are listed. The presence of elements in an alloy from the second column, ‘±,’ can have both positive and negative effects on the general cladding material properties, especially on its resistance to corrosion phenomena. For example, Fe has a positive effect on lattice matching but can increase the hydrogen pick-up. Elements from the last column accelerate corrosion or did not demonstrate any influence on the corrosion rate.
Table 2. Elements present in Zr alloys.
Some of the elements from can be part of the second-phase particles (SPP) or solute content with a pronounced effect on corrosion [Citation10, Citation13, Citation63, Citation67, Citation68], see below.
3.2 Impact of alloy microstructure on the oxide microstructure formation
The numbers of cycles during the growth of the ZrO2 layer, duration of individual cycles, transition points, and rate of oxidation within a cycle are different from the alloy microstructure and its chemical composition, . Such. the study of Brossmann et al. [Citation69] specified that the volume diffusivity of oxygen in undoped m-ZrO2 is, in comparison to the melting temperature, much higher than in other transition metal oxides but significantly lower than in Y-, Ca-, or Mg-stabilized ZrO2, where the oxygen diffusion is controlled by the migration of structural oxygen vacancies. Adamson et al. [Citation17] indicate that the accumulation of alloying elements (e.g. Nb in Zr-2.5%Nb alloy) and impurities at the ends of columnar oxide crystallites disrupt the growth of columnar grains: instead of forming ZrO2, NbO2 is formed. The influence of alloying elements on the cyclic oxidation kinetics, crystallographic structure of the protective layer, and the nanoscale pore formation was studied by Gong et al. [Citation70] for N18 (Northwest Institute for Non-ferrous Metal Research, China), M5® alloys (AREVA, France) and three Zr–Nb–Y ingots: Zr1.0NbxY (x = 0, 0.1, 0.4 wt.%), .
Figure 7. Corrosion film thickness as a function of exposure time in 360°C pure water for various zirconium alloys as indicated [Citation37].
![Figure 7. Corrosion film thickness as a function of exposure time in 360°C pure water for various zirconium alloys as indicated [Citation37].](/cms/asset/9a2b86f7-8c7c-4eb0-880c-2fe1080f1a54/tnst_a_2127954_f0007_b.gif)
Table 3. Chemical composition of the alloys investigated in [Citation70] (wt.%).
The oxide films on the M5® alloy displayed one integral layer, while two layers were found on the N18 alloy. In the case of the Zr1.0NbxY alloys, the number of periodic layers was increased with increasing Y content, .
Figure 8. Cross-sectional morphology of the oxide films formed on zirconium alloys: (a) M5®, (b) N18, (c) Zr1.0Nb, (d) Zr1.0Nb0.1Y, and (e) Zr1.0Nb0.4Y. From [Citation70].
![Figure 8. Cross-sectional morphology of the oxide films formed on zirconium alloys: (a) M5®, (b) N18, (c) Zr1.0Nb, (d) Zr1.0Nb0.1Y, and (e) Zr1.0Nb0.4Y. From [Citation70].](/cms/asset/75d5d28f-3f62-422d-a9f1-bbe0eee1d2df/tnst_a_2127954_f0008_b.gif)
The oxide films studied in [Citation70] mainly consisted of columnar grains aligned parallel to the growth direction of the oxides. No oxide pores were observed for the tightly aligned columnar oxide grains formed on the Zr1.0Nb alloy. Unlike that, the nanoscale spherical pores in strings were observed at grain boundaries of the columnar oxides formed on the Zr1.0Nb0.1Y and Zr1.0Nb0.4Y alloys: the diffusion of yttrium from the Y SPPs into cation sub-lattices of the oxides generates the nanoscale pores.
Wei and coworkers [Citation33] investigated three recrystallized Zr–Sn–Nb alloys (ZIRLO®, A-0.6Sn, and A-0.0Sn) with tin concentrations ranging from 0.01 to 0.92 wt.% at 360°C and 18 bar in simulated primary water to clarify the mechanism that leads to the transition of the corrosion rate. It was demonstrated that all tin present in the alloy passes into the oxide as corrosion progresses, which may then influence the nature of the oxide. The onset of transition was delayed from as early as 140 days in ZIRLO material to as late as more than 360 days in A-0.0Sn material. The actual corrosion kinetics in the pre-transition stage was practically identical for all studies of alloys. The differences in the transition kinetics were explained by the influence of the alloy chemistry on the tetragonal phase stabilization. The amount of tetragonal phase present and, more importantly, the level of tetragonal-to-monoclinic phase transformation both decrease with decreasing tin levels, suggesting that tin is a tetragonal oxide phase stabilizing element. Studies [Citation53, Citation65, Citation71, Citation72] also reported that the higher the content of Sn in the Zry-4, the higher the content of the tetragonal oxide. Authors of [Citation33] concluded that corrosion resistance was improved significantly with decreasing tin content, i.e. tetragonal phase, which is in some discordance with other investigations, but following an ambivalent nature of a tetragonal oxide pointed out earlier.
Recently Renčiuková et al. [Citation30] studied the corrosion rate of zirconium alloys with a focus on the terms of transition and cyclic nature of the kinetics. The study is performed based on the electrochemical nature of the process.
Two zirconium alloys, Zry-4 and Zr1Nb, , were exposed to a non-active VVER primary coolant (de-aerated by argon or nitrogen solution contained 1050 ppm B as H3BO3, 15.9 ppm K as KOH and 1 ppm Li as LiOH, dissolved oxygen below 5 ppb) at 340°C for almost 700 days. Corrosion kinetics was mainly defined using oxide resistance and polarization resistance. The studied in [Citation30] alloys differ in time to the
Table 4. Chemical composition of the measured in [Citation30] Zr alloys.
start and the end of the transition, critical oxide thickness, and the number of kinetic cycles, : the Zry-4 gradually went through three transitions and Zr1Nb went through only one transition and has a lower total oxide thickness than the Zry-4 alloy and did not have visible cracks perpendicular to the oxide surface. The first transition of Zry-4 and Zr1Nb started at 183d and 396d, respectively. The transition was a little longer in the case of Zry-4 (80d) than Zr1Nb (62 d). The possible evolution of the oxide is common for both alloys [Citation30]. At the same time, alloying elements affect the pre-transition and transition kinetics of Zr-Nb and Zr-Sn type alloys differently [Citation30, Citation71]. The significant decrease of the polarization resistance at transition for the Zry-4 was explained in [Citation30] by the ambivalent nature of a tetragonal oxide: higher content of t-ZrO2 increases the corrosion rate at the transition through a tetragonal-to-monoclinic transformation but also increases the oxide film resistance in the pre-transition.
Figure 9. Time evolution of the oxide resistance, Rox, and polarization resistance, Rp, of Zry-4 (a) and Zr1Nb (b). From [Citation30].
![Figure 9. Time evolution of the oxide resistance, Rox, and polarization resistance, Rp, of Zry-4 (a) and Zr1Nb (b). From [Citation30].](/cms/asset/4250a430-9f83-4536-8104-44e83fdcf68f/tnst_a_2127954_f0009_oc.jpg)
It is recognized in [Citation30] that the kinetic law of growth of the new barrier oxide was similar to the barrier oxide layer at the pre-transition, but the second and third kinetic cycles of the Zry-4 alloy were different from the nature of the first cycle oxide where the resistance was irrecoverably affected by the presence of a porous part of the oxide, and the alloy gradually went through three shortening kinetic cycles.
3.3 Influence of SPPs on the oxide microstructure formation
SPPs impact corrosion as possible cathodic sites for the corrosion reaction; as possible short-circuit paths through the protective oxide, either on their own or due to nearby cracking; as possible places for hydrogen storage; and as possible sources of stress in the oxide layer upon delayed oxidation. Also, SPPs may retard or accelerate the process of recrystallization: SPPs create additional interfaces within the matrix and through that modify the grain structure and crystallographic texture after recrystallization. For more information, see, please, studies [Citation10, Citation44, Citation60].
The second-phase particles in Zircaloy-4 and ZIRLO™ were investigated by De Gabory et al. in [Citation39]. They found that, in Zircaloy-4, oxidized Zr(Fe, Cr)2 precipitates appear as a collection of small-equiaxed grains with a diameter of the order of 10 nm, separated by small cracks, with a high concentration of tetragonal oxide. In ZIRLO™ samples two different types of second-phase particles are observed: ZrFeNb particles, similar to the structure of Zr(Fe, Cr)2 precipitates, and β-Nb precipitates, which are roundish and smaller in size. The precipitates are incorporated into the oxide in their metallic state and exhibit a crack on the side opposite the oxide/metal interface. While the Zr(Fe, Cr)2 remains unoxidized (and its volume does not change), the matrix around it keeps oxidizing and expands in the oxide growth direction, leading to the formation of a crack on top of the precipitates. The difference with Zircaloy-4 is that when ZIRLO™ precipitates oxidize, they become amorphous. The oxidized precipitates show no crack on top. Authors of [Citation39] concluded that as porosity develops at oxide grain boundaries the smaller oxide grains in Zircaloy-4 than in ZIRLO™ should lead to greater oxide porosity. Further, the porosity would be more likely to develop on higher-angle grain boundaries than in well-aligned grains: Zircaloy-4 has a greater degree of misorientation in the oxide than ZIRLO™.
Cox in his study [Citation6] pointed out that galvanic potentials can appear between large SPPs (or clusters of SPPs) and the Zr matrix, which consequently leads to the galvanic currents between SPPs and the Zr matrix and therefore greatly increases the electronic conduction of zirconia and corrosion rate.
Another effect was investigated in [Citation73]. It was shown that a surface dissociation of oxygen molecules on particle surfaces bearing Fe, Ni, and Cr (Zr2Fe, Zr2Ni, ZrCr2) is far higher than a dissociation on the surface Zry-2. Such a catalytic effect of SPPs results in an increase in the corrosion rate and in a reduction in the rate of hydrogen penetration into the metal through the oxide film.
The doping of oxide with elements having lower valences compared to that of zirconium (e.g. iron and chromium) can lead to the generation of anion vacancies at the metal/oxide surface. Oxidation of the intermetallic results also in volume expansion and the surrounding columnar ZrO2 is damaged by the high local stresses [Citation74].
4 Electrochemistry of Oxidation kinetics
How it follows from the above that corrosion is controlled with an integrated effect of diffusive-electrochemical processes: molecular, electron, and ion transfer through the metal, oxide layer, and surrounding medium; electrochemical/chemical reactions on the metal/oxide, metal/environment interfaces and in metal, metal oxide, and the surrounding medium, . The start point of corrosion is defined by the electrochemical corrosion potential determined, in an ideal case, by the electrical, chemical, and thermodynamical properties of the alloy. In a real case, the difference between the chemical potentials of alloy and oxide, molecular and ion concentration gradients on the metal/environment, and metal oxide/surrounding medium interfaces (the diffusion polarization), fluid velocity, etc., should be accounted for.
Figure 10. Electrochemical processes are involved in the ‘electro-chemical corrosion’ phenomenon.
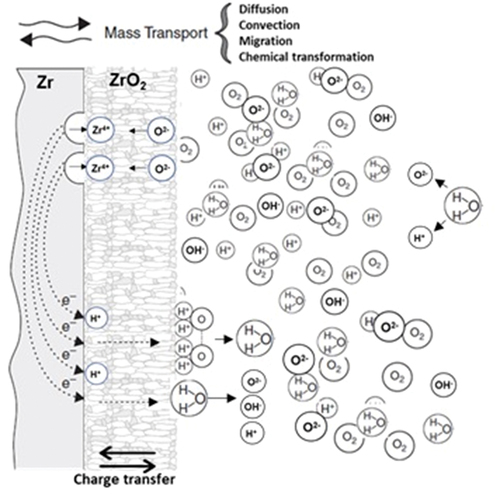
Electrochemistry is a fundamental part of corrosion science, which integrate electrochemical and quanta-physical processes [Citation16, Citation75, Citation76]. Surprisingly, despite the comprehensive theoretical investigations and attempts to describe corrosion on the molecular level [Citation2, Citation6, Citation65, Citation75, Citation77–79] there is a big deficit in the literature on the investigation of electrochemistry of Zr corrosion.
4.1 Anodic polarization curve
Polarization curves (PC) are used extensively to show how the factors listed above and mentioned in the introduction determine the corrosion potential and corrosion current density, different characteristics important in understanding the corrosion behavior of the alloy [Citation8, Citation77, Citation79] and protective layer formation.
The anodic PC for a most general case of film formation/destruction is schematically shown in . Each mode of corrosion is started at a specific potential coupled with a defined current density (corrosion rate), : the active corrosion starts with the corrosion potential Ecorr, where the metal enters the ionic state; it is followed by the formation of the passive layer with reducing current density jp; passive state mode with the stable current density, jsp; film breakdown with re-passivation, pitting corrosion and stress corrosion cracking; transpassive dissolution mode of an oxide layer with the rate of corrosion reaching the maximum values. Transpassivation is a rapid, catastrophic, dissolution process of a passivated metal, without the possibility of a re-passivation.
Figure 11. Schematic potential–current curves for metallic passivation, passive-film breakdown, pitting dissolution, and transpassivation; Ecorr is the corrosion potential; EP is the passivation potential; ESP is stable passivation potential; Eb is the film breakdown potential, Epit is the pitting potential, and ETP is the trans passivation potential, dash black line – cathodic curve. Each potential is coupled with corresponded current density. Extended Figure1.6 from [Citation79].
![Figure 11. Schematic potential–current curves for metallic passivation, passive-film breakdown, pitting dissolution, and transpassivation; Ecorr is the corrosion potential; EP is the passivation potential; ESP is stable passivation potential; Eb is the film breakdown potential, Epit is the pitting potential, and ETP is the trans passivation potential, dash black line – cathodic curve. Each potential is coupled with corresponded current density. Extended Figure1.6 from [Citation79].](/cms/asset/6b64247b-d4c8-438c-82dc-dfa80364b5c9/tnst_a_2127954_f0011_oc.jpg)
The response of an alloy to a corrosive environment is highly individual, therefore the curves differ greatly in shape and position for different alloys. Alloying elements and SSPs influence chemical potentials and thereby electric fields in metal/oxide areas and type of conduction [Citation30]. The reaction mechanisms on electrodes (Equations (2.1)-(2.8)) depend on the kinds and amounts of alloying elements, the presence of impurity atoms, and SSPs in the oxide lattice. The reliable anodic PC of alloys is very useful for the evaluation of the corrosion current.
The nature of the solution, the H+ concentrations, and the temperature are operating conditions that strongly influence the anodic polarization curves and the passive region. For example, at increasing the oxidizer concentration, the cathodic potential–current curves (dash black line in ) can cross the anodic potential–current curve at different points, i.e. with different Ecorr and icorr. So, an unstable passive state can arise if the cathodic curve crosses the curve at two potentials, one in the passive state and the other in the active state.
Chen et al. [Citation80] investigated the reduction of the passivation region in the Zr/H2O system with temperature increase at PWR conditions (i.e. pH = 6.9–7.4, 360°C). The passivation region will have a very narrow pH range due to the promoted corrosion processes at high temperatures [Citation80]. These results were presented by the authors of [Citation80] with the Pourbaix diagram, .
Figure 12. Potential-pH diagram (Pourbaix) diagram of zirconium in water at elevated temperature a) at 298 K b) at 333 K c) at 373 K d) at 423 K [Citation80].
![Figure 12. Potential-pH diagram (Pourbaix) diagram of zirconium in water at elevated temperature a) at 298 K b) at 333 K c) at 373 K d) at 423 K [Citation80].](/cms/asset/ef8f0ac4-56b7-4721-9bc9-eea8bc9675d7/tnst_a_2127954_f0012_b.gif)
Mamun et al. [Citation81] investigated the Zirconium passivation and passive film breakdown during anodic oxidation of zirconium (purity of 99.5%) in aqueous systems with particular attention to the passive and transpassive regions using cyclic voltammetry (CV). Mechanically polished zirconium electrodes have been oxidized anodically in pH = 8 buffer solutions containing 0.021 M chloride, with varying sulfate concentrations. They have shown that the characteristics of the polarization curve and the potential range of the Zr passive layer were changed significantly by variation of the solution chemical content, , while the pitting in some cases was completely suppressed. The measured polarization curve had a sharp increase marking the beginning of the transpassive region, , the breakdown of the passive film and oxygen evolution at higher potentials contributed to the measured current.
Figure 13. (a) Anodic oxidation of Zr in pH = 8 buffer solution containing 0.021 M Cl− and different sulfate concentrations [Citation81]; b) Potentiodynamic polarization curves on Zr electrode in aerated phosphate buffer solutions of various pH values (scan rate: 10 mV s-l): (1) 11.0, (II) 9.0, (III) 7.0, (IV) 5.0, (v) 2.5, (VI) 1.6 and (VII) 0.9 pH, [Citation82].
![Figure 13. (a) Anodic oxidation of Zr in pH = 8 buffer solution containing 0.021 M Cl− and different sulfate concentrations [Citation81]; b) Potentiodynamic polarization curves on Zr electrode in aerated phosphate buffer solutions of various pH values (scan rate: 10 mV s-l): (1) 11.0, (II) 9.0, (III) 7.0, (IV) 5.0, (v) 2.5, (VI) 1.6 and (VII) 0.9 pH, [Citation82].](/cms/asset/7e9e195a-ae7f-48f9-893b-044be424a075/tnst_a_2127954_f0013_b.gif)
The change of the zirconium passivation potential with a decrease in the pH solution was also detected in the study of Abdel Rahim et al. [Citation82], .
There is no comprehensive scientific definition of transpassive corrosion in the literature, but general ideas or statements of some facts, e.g.: transpassivation is a phenomenon in which a passivated metal starts rapid dissolution if the electrode potential of the metal becomes too positive; transpassive dissolution of a range of alloys is closely related to passive film breakdown and localized corrosion phenomena; transpassive dissolution of a metal or alloy can be defined as the formation of chemical species in a valence state higher than that in the primary passive film formed on the material. The elucidation of the transpassive regime of corrosion, to our knowledge, was more clearly done by Sato [Citation79]: in the transpassive potential range, the interfacial potential between the film and the solution is not constant but depends on the anodic potential of the metal. The passive film is stable as long as the Fermi level (the electrode potential) of the metal anode is within the bandgap of electron energy between the conduction and valence bands of the film, a situation that realizes the nonmetallic nature of the interface and hence makes the interfacial potential independent of the metal potential [Citation79]. As the anodic potential increases, the Fermi level finally reaches the valence band edge of the film at the film–solution interface, and the quasi-metallization (electronic degeneracy) is realized at the same. The film–solution interfacial potential, hence, turns out to be dependent on the anodic metal potential, and as a result, potential-dependent transpassive dissolution occurs beyond the transpassivation potential, ETP., . Goossens et al. [Citation75] developed the band model for passive films on Zr and applied a similar elucidation for the transpassive state.
Unfortunately, the data available do not permit a comparison of the Zr alloys polarization curves for different operating conditions. Although the potential–current curve for metallic passivation has evidence connection with the cyclic type of Zr oxidation and oxide multilayer formation, it is almost impossible to find in-depth investigations of the electrochemical corrosion potential of Zr and the anodic PC for Zr alloys. It is even more surprising because it is intuitively clear that regular repeating structures of the Zr oxide layer are determined first with fundamental molecular processes and only secondary with stochastic processes of cracks and pore formations.
The transpassive corrosion of ZrO2 was faintly examined, is least investigated, and has a rudimentary understanding unlike the transpassivation mechanism of stainless steel, which thanks widely used in many industrial processes is mostly of scientific and practical interest [Citation27, Citation79, Citation83–86].
4.2 Possible galvanic effects
The coupling of different metals or alloys causes the corrosion potentials and corrosion current densities to change, frequently significantly, from the values for the two metals in the uncoupled condition. The magnitude of the shift in these values depends on the cathodic and anodic reactions, solution resistance, and the relative magnitude of the areas of the two metals.
Cox [Citation6] analyzed possible galvanic effects, which can arise in the in-reactor conditions. The irradiation was increasing the conductivities of the Zr oxide films to the point where galvanic effects became possible, i.e. galvanic current can be passed for a given potential difference between dissimilar metals. Ramasubramanian [Citation87] observed the coupling of the two materials in the gap via ionic transport in the coolant. This was facilitated by the potential difference between the two metals, the water chemistry, and the transport of species under their concentration gradients.
The effect of the SPP size on the susceptible batches of cladding may again affect the electronic conductivity of the oxide since small precipitates dissolve most rapidly during irradiation [Citation6].
Cox [Citation6] also pointed out that CRUD deposits on zirconium oxide surfaces can have a big effect on the electronic conductivity of the oxide. The effect depended on the matching of the band gaps of the chemical form of crud deposits in contact with the zirconia surfaces with the bandgap of the ZrO2. The galvanic effect is responsible for the well-known phenomenon of shadow corrosion [Citation17, Citation88–90]. That is especially pronounced in BWR reactors, where the dissimilar metal elements are collocated rather narrowly in the core, .
Figure 14. a) BWR control cell with four bundles and a control-blade [Citation88]: 1 – corrosive shadow; 2 – control blade; 3 – fuel channel; 4 – gap; b) Photo of shadow corrosion on the surface of the BWR fuel channel [Citation89].
![Figure 14. a) BWR control cell with four bundles and a control-blade [Citation88]: 1 – corrosive shadow; 2 – control blade; 3 – fuel channel; 4 – gap; b) Photo of shadow corrosion on the surface of the BWR fuel channel [Citation89].](/cms/asset/47f6331d-c04c-4fea-8621-344edbacdaea/tnst_a_2127954_f0014_b.gif)
In VVER and PWR shadow corrosion is not observed, but this does not mean that galvanic corrosion does not act. Since the VVER core contains metal structural elements with different electrochemical potentials, the presence of electrical contact between them or through an electrically conductive solution should lead to galvanic effects. The distance between these structural elements in VVER is relatively large; therefore, the effect of galvanic corrosion should be distributed relatively over a large surface, and the contribution of this type of corrosion to the overall corrosion of fuel elements in VVER can be significant [Citation90].
5 Oxidizing medium
The chemistry of coolant is defined by the interaction of five main chemical systems [Citation91]:
chemical content of the reactor coolant with different chemical additives such as boric acid and lithium hydroxide (H3BO3 and LiOH), KOH, hydrogen, oxygen, halogenides, sulfates, chromates, nitrates, etc.
water radiolysis
the possible escaped corrosion products and their hydroxides and aqueous ions
dissolution of Zr oxides and hydroxides
deposition of particulate and colloidal metal oxides, dissolved and hydrated metal ions (CRUD)
These systems are involved in various processes of coolant: heterogenic and catalytic (due to the influence of intermetallic inclusions) reactions on the different surfaces, in porous oxides; heat and mass – transfer with different scales, influenced by temperature, pressure, and irradiations; adsorption-desorption of gas/liquid medium molecules and oxide film; corrosion process, etc.
A major focus of the control of chemistry parameters is the mitigation of corrosion, a problem that affects power plants universally [Citation91]. Despite the vital and fundamental importance of coolant chemistry, the ample amount of published studies/information, and the strict control standards [Citation8, Citation47, Citation48, Citation92–97], water chemistry parameters are studied also mostly empirically. There are not any complete detailed or at least semi-detailed well-calibrated chemical reaction schemes in the open literature, which describe all interacting chemical and electrochemical processes in the coolant.
5.1 The chemical content of the reactor coolant
The multitasking of the reactor coolant is operated with different chemical additives such as boric acid and lithium hydroxide (H3BO3 and LiOH), with the addition of hydrogen, oxygen, halogenides, sulfates, chromates, nitrates, etc. During power operation, the coolant is maintained slightly alkaline and reducing, is in a subcritical state (290°C–330°C and ~16 MPa) and acts as a highly corrosive medium. There are different requirements for water depending on the reactor type. , following from [Citation9, Citation98] demonstrates the typical reactor environments to which the zirconium alloys are exposed.
Table 5. The typical coolant specifications [Citation9, Citation98].
Boric acid, H3BO3, is usually added as a neutron absorber, while with lithium hydroxide, and LiOH, the pH value of the reactor coolant is controlled. Mesmer et al. [Citation99] developed the model for the estimation of the pH for any given temperature, [B] and [Li] with a reaction set:
It was used in studies [Citation27, Citation100] to follow the oxide solubility and metal corrosion rate dependence on pH.
H3BO3 and LiOH readily influence the corrosion rate of the system materials. An increase in the corrosion rate of 10–30% has been observed when the maximum coolant lithium content was increased from 2.2 to 3.5 ppm [Citation66], the effect of higher lithium concentrations being much more pronounced [Citation101].
The key factors of the LiOH effects on corrosion, which may significantly degrade the barrier layer [Citation102], are modification of crystal growth of equiaxed grains [Citation103], hydrogen absorption [Citation93], and pore formation due to preferential cubic or tetragonal ZrO2 dissolution [Citation92]. The acceleration of hydrogen absorption in the LiOH water was ascribed to the formation of degraded or open grain boundaries up to locations very near the metal/oxide interface [Citation93, Citation104]. Besides that, Li builds different chemical compounds with boron which further can be precipitated within the deposit (see below).
Reducing conditions of coolant are operated by dissolved hydrogen concentrations that are kept high enough, , to suppress dissolved oxidants (OH, O, O2, and H2O2) to very low levels by radiation-induced reactions. To produce the specified hydrogen concentration, the NH3 can be injected into the coolant. Also, for the chemical scavenging of O2 during start-up, hydrazine can be injected into the coolant: N2H4 + O2 = N2+ H2O [Citation105]. Thereby ammoniated water showed a lower impact on corrosion rate at the reactor temperature, but nitrogen chemistry is investigated mostly for temperatures higher than 800°C, where nitrogen chemical reactions are more important [Citation106].
Bojinov et al. [Citation107] indicated that both rate constant of Zr oxidation at the alloy/film interface and the diffusion coefficient of oxygen vacancies in the barrier layer increase, whereas both the defect-induced resistivity of the inner layer and the overall resistivity of the outer layer decrease with increasing KOH concentration.
In the paper [Citation66] the effect of LiOH, KOH, and fluoride concentration on the kinetics of oxidation and the conduction mechanism of the formed oxide has been further assessed, and the main kinetic and transport parameters are estimated by fitting the model equations to the measured data. It is shown that the increasing LiOH, KOH, and F content in the coolant decreases the oxide barrier thickness, and this effect is more pronounced at the highest concentrations of the respective additives (10 ppm LiOH, 56 ppm KOH, and 1000 ppb fluoride). A possible explanation for this observation is that the increase in LiOH, KOH, and fluoride concentration leads to an increase in the solubility of zirconium oxide at its interface with the electrolyte.
To reduce radiation fields and corrosion in the primary system, and to prevent/mitigate the crud deposition in the core, zinc injection into the reactor coolants is applied [Citation108–110]. Zinc due to its higher substitution potential [Citation109, Citation111] can substitute the radioactive ingredients of the corrosion oxide layers and discharge them doing the protective layer more stable.
The available theoretical tools to predict the corrosive conditions have an unacceptable level of uncertainty and, in the practice, several instruments are available to indicate corrosion by monitoring the key parameters compiled in [Citation91]. But such measurements are not direct, there are additional requirements to interpolate or extrapolate the data to locations and parameters where the measurements cannot be taken. Some radiolytic species are so unstable that determining their distribution and the resulting electrochemical corrosion potential throughout the cooling systems can only be achieved with extrapolation based on theoretical tools.
5.2 Radiolysis, corrosion products, and aqueous species
The different oxidation and corrosion products that escape to the water are dissolved in the water and are carried through the system [Citation91, Citation100, Citation105]. The migration of released products from place to place within the system involves a complex set of chemical and physical interacting mechanisms leading to the formation of hydroxides, aqueous species, and ions [Citation100, Citation105]. The formation/transformation of these different chemical compounds in the primary coolant is impacted by active radicals and ions resulting from the water radiolysis.
Radiolysis of steam/water mixture or water solution can take place when exposed to high-energy particles (neutrons, alpha particles, gamma quanta, and fission fragments) breaking the bonds between atoms or exciting the molecules thereby triggering further reactions.
The further change in concentrations of molecules, radicals (H, OH, О, НО2 and Н2О, Н2, О2, Н2О2), their ions, and active particles can be simulated with corresponding chemical kinetic reaction mechanisms [Citation112]. One example of them is a model used in [Citation100, Citation105], . The rate constant, kj (j denies the reaction number in ) is a function of coolant temperature described with Arrhenius’ law.
Table 6. The reaction set is used in the radiolysis model of Macdonald et al. [Citation100], and Betova et al. [Citation106].
where ko is the rate constant at temperature To, Ea is the activation energy, R is the universal gas constant, and T is the temperature in Kelvin.
In the studies [Citation73, Citation113], it is shown that for the coolant in the vapor phase, the radiolysis crucially affects the oxygen-and-hydrogen bearing radicals whereas, for the coolant in the liquid phase, the coolant chemical composition plays a governing role in a setting-up concentration of oxygen and hydrogen radicals at temperatures over 300°С.
The chemical and mechanical interactions of the passive layer with active species in the coolant lead to the transformation of the barrier layer into a non-protective porous layer, the dissolution/precipitation of corrosion products, and the change of passive layer thickness [Citation66, Citation114, Citation115].
5.3 Solubility of Zr oxides and hydroxides in water solutions
Nishino et al. [Citation116] showed that the water radiolysis species accelerated the dissolution of the oxide film, which generated the microcracks at the oxide film. That caused the t- to m-ZrO2 transformation, i.e. lower tetragonal-ZrO2 fraction. The authors proposed a possible mechanism based on surface reactions of OH− leading to crack initiation and propagation, . The lowest solubility of ZrO2 was evaluated at about pH 6.6 in 25°C water. The solubility was larger at both lower and higher pH.
Figure 15. Model of dissolution process of zirconium oxide into water, from [Citation116].
![Figure 15. Model of dissolution process of zirconium oxide into water, from [Citation116].](/cms/asset/e4b82060-55af-4713-b92c-4c8a792a1917/tnst_a_2127954_f0015_b.gif)
The possible reactions of electrochemical and chemical dissolution of possible dissolved metal oxides and different corrosion products proposed by Macdonald and coauthors [Citation100] are collected in .
Table 7. Reactions describing the dissolution of corrosion products into the primary coolant [Citation100].
The main problem occurring by modeling reactions in the coolant is the evaluation of the reaction rate constants, electrochemical corrosion potential, and solubility values, thermodynamic properties of elements in aqueous solutions. These vital data are poorly constrained, published values are frequently contradictory, may vary by several orders of magnitude, and should be regarded as being little more than rough estimates or generally not known [Citation107, Citation112, Citation117–121].
Therefore, we found it appropriate to present here useful information from [Citation121–124].
Preformed by Duro et al. [Citation121] analysis of the hydrolysis system of Zr includes species Zr(OH)3+, Zr(OH)4(aq), Zr(OH)5 – and Zr(OH)6 2 – and some selected reaction for the zirconium hydrolysis, . Also, the authors of [Citation121] considered the precipitation of two different amorphous Zr hydroxides: Zr(OH)4(am) and Zr(OH)4(s) as solubility limiting phases. E. Curti and C. Degueldre [Citation123] studied hydrolysis constants for species Zr4+, ZrOH3+, Zr(OH)4, and Zr(OH)5−. In the paper [Citation122] the solubility of the different Zr oxides and hydroxides are analyzed and gathered in the form of . Kritskiy A. [Citation124] evaluated thermodynamic data related to the ZrO2 and Zr4+ dissolution, , and solubility dependence of ZrO2 on pH and T. It was shown that Zr4+ reacts very fast and Zr(OH)4 dominates for all pH = 1–9.
Table 8. Selected data for the Zr hydrolysis scheme from [Citation121] β0 -stability constant, KS -equilibrium constant.
Table 9. Solubility of Zr oxides and hydroxides in water and water solutions at 25°C [122]. The exact full references can be found in [122].
Table 10. Thermodynamical properties of hydroxo complexes of Zr at 25°C [Citation124].
5.4 CRUD
The products, (R1)-(R8) in the form of particulate and colloidal metal oxides along with dissolved and hydrated metal ions (so cold CRUD) are deposited on the fuel cladding surfaces where they reside long enough and are responsible for corrosion acceleration (as in the crud-induced localized corrosion, or CILC, of the BWR fuel sheaths) and for fouling of the fuel. In modern high-duty PWRs, such fuel fouling can cause CIPS (crud-induced power shifts – also known as AOA, axial offset anomaly) as boron is sequestered in deposits and affects the neutron balance [Citation103]. Boron can be precipitated in CRUD pores as the meta-borate (LiBO2) [Citation116], as LiB(OH)4 [Citation109] or as nickel-iron borate (Ni2FeBO5) [Citation116, Citation125–128].
from Iva Betova et al. [Citation105] illustrates qualitatively the CRUD deposition and influences the temperature decrease on the CRUD. With this illustration, we wanted to demonstrate the complexity and another side of the physicochemical processes on the oxide/coolant interface influencing the corrosion.
Figure 16. (left) CRUD structure with severe AOA, (right) transformation of the CRUD after temperature reduction in a subsequent cycle, from [Citation105].
![Figure 16. (left) CRUD structure with severe AOA, (right) transformation of the CRUD after temperature reduction in a subsequent cycle, from [Citation105].](/cms/asset/19877cac-b177-432b-aa9f-cd450af6095b/tnst_a_2127954_f0016_oc.jpg)
In the case of the presence of the crud layer on the alloy surface, coolant boiling is possible in the pores of this layer. This results to a great extent in an increase in the relative fraction of the vapor phase in the coolant. Heterogeneous radiolysis can take place in the oxide film pores (at the post-transition stage of oxidation) and in the crud layer pores – linear losses of irradiation energy are higher in a solid body than in the liquid and gas phase [Citation113].
The chemical model with a strongly evaluated valid parameter range, besides theoretical meaning, has very important practical significance. Lister & Uchida [Citation91] underlined the major aims of theoretical efforts that should be done for coolant chemistry:
Theoretical models for water radiolysis;
Theoretical and empirical models for crack propagation rate;
Theoretical evaluation of pH of cooled water;
Theoretical oxidation models for characterization of the oxide film.
Another problem pointed out by Lister & Uchida [Citation91] is the fact, that although most of the phenomena are understood internationally, the details of chemistry control remain specific to individual industry groups [Citation110]. Technical guidance for standardizing chemistry control in the nuclear industry internationally, like those available for the fossil-fueled power industry, would be very useful.
6 Hydrogen Pickup
6.1 General remarks
The corrosion of the cladding tube is associated with hydrogen ingress into the zirconium cladding (commonly referred to as hydrogen pickup) [Citation37, Citation129]. The hydrogen pickup can lead to the accumulation of hydrogen in the alloy, the formation of brittle hydrides, and zirconium hydride platelets, which significantly reduce the ductility of Zr alloys and can have a deleterious effect on the Zr corrosion [Citation15, Citation130].
In addition to the hydrogen initially contained in the zirconium cladding and the fuel pellets, the main external hydrogen sources are cathodic-generated H on the oxide layer/water boundary, Equations (2.3)-(2.8), the radiolysis dissociation of the coolant water, and the H2 gas dissolved in the primary coolant. It has been speculated that the hydrogen enters the oxide in the form of protons [Citation11, Citation129] and may pass through the oxide barrier layer [Citation10]: via local imperfections (cracks and pores) in the barrier oxide film; through non-oxidized SPP (e.g. containing Ni or Cu), acting as windows; by the hydrogen diffusion in the oxide grain boundaries and oxide cracks. The drivers of hydrogen flux from the outside of the oxide layer to the metal–oxide interface are an electrical field and the H concentration gradients between oxide/coolant and metal/oxide interfaces [Citation15]. In the alloy, the Fickian flux drives hydrogen from the outside of the cladding tube inwards, the Soret flux goes in the direction of decreasing temperature. Since hydrogen is highly mobile in zirconium alloys [Citation15], it can respond reasonably quickly to temperature and stress gradients that drive it to precipitate preferentially in the regions of high hydrostatic tensile stress, low temperature, oxide film spallation, or it can accumulate in the interpellet region where the heat flux is lower [Citation10, Citation15, Citation131, Citation132]. The absorbed hydrogen atoms diffuse into Zr metal as long as its concentration remains below the terminal solid solubility (TSS) of hydrogen for the zirconium alloy (80–150 ppm, depending on temperature) [Citation131, Citation133].
Various investigators estimate that 30–100% of the hydrogen produced in the cladding corrosion can be absorbed [Citation134, Citation135]. The hydride at the corroding interface is a two-phase mixture of zirconium metal and hydrides. Below 773 K their compositions may slightly deviate into [Citation135, Citation136]: γ-Zr, with H in solid solution in the matrix; δ-hydride, ZrH1.5 to ZrH1.66, which initially precipitates as platelets in α-Zr; ε-hydride, fct, ZrH1.66 to ZrH2, which comes from a martensitic transformation of δ-hydride. Through the changes in hydrogen atom distribution hydrides may preferentially precipitate in a so-called ‘hydride rim’ near the outer cladding surface or hydride blisters [Citation15, Citation131].
The faction of the total hydrogen that gets picked up by the cladding is called total hydrogen pickup fraction, defined usually as
and
are the amount of hydrogen absorbed in the cladding and the amount of hydrogen generated/absorbed on the oxide/coolant from the start of corrosion exposure, respectively. The
depends strongly on the alloying element additions, the material crystallographic texture and the microchemistry, and the corrosion conditions [Citation11, Citation15].
The additive elements (e.g. Fe, Cr, Nb, Sn, and Ni) in the Zr alloys may either behave like trapping sites or directly decrease the hydrogen diffusivity in the oxide or they may form second phase precipitates that have been hypothesized to be a preferred path for hydrogen migration or a source for pores or cracks formation [Citation137].
Couet et al. [Citation138] presented measuring hydrogen pickup fraction for Zircaloy-4, ZIRLO®, Zr–2.5Nb, Zr–2.5Nb–0.5Cu, and three model alloys: pure Zr, Zr–0.5Cu and Zr–0.4Fe–0.2Cr. The study was focused on the investigation of the effects of alloying elements, microstructure, and corrosion kinetics on hydrogen uptake. The results shown that hydrogen pickup fraction varies significantly with exposure time and between alloys. That was indicated that Nb reduces hydrogen pickup fraction whereas increases it, [Citation138]. It was proposed that Nb can be oxidized as Nb2O5 at the oxide/water interface and acts as a local donor reducing the hydrogen before absorption of the protons in the oxide layer. The reasons why Cu addition increases hydrogen pickup fraction compared to other alloys with a similar oxide thickness are yet to be determined. That was shown [Citation138] that alloys with larger size of Zr(Fe,Cr)2 has a lower compared to the alloy with smaller Zr(Fe, Cr)2 precipitates. It was also pointed out that the oxide electronic conductivity plays a key role in the hydrogen pickup mechanism. If the metallic precipitates enhance the charge transport and, consequently, electronic conductivity of the oxide layer, that will in turn reduce the hydrogen pickup.
Figure 17. Scheme of the influence of the alloying elements on oxidation kinetics as a function of the exponent n from the power law fit of the weight gain wg = ktn and hydrogen pickup fraction for various zirconium alloys, from [Citation138].
![Figure 17. Scheme of the influence of the alloying elements on oxidation kinetics as a function of the exponent n from the power law fit of the weight gain wg = ktn and hydrogen pickup fraction fHt for various zirconium alloys, from [Citation138].](/cms/asset/ffd03ab6-6ddf-4b96-8a2a-8c0e6ee24855/tnst_a_2127954_f0017_b.gif)
Motta et al. [Citation11] pointed out that alloying additions have a profound impact on the hydrogen pickup mechanism except for Ni and Cu which increase pickup, and Sn, whose effect is not well determined. Authors of [Citation11] underlined that the effects of precipitate size and volume fraction are not well understood, but for a given precipitate volume fraction, an increase in the number of precipitates (and thus a reduction of their sizes) increases , possibly because more cathodic sites are available.
In the recently published review of Arthur T. Motta et al. [Citation15], the processes of hydrogen pickup and the ultimate impact of the hydrides on cladding mechanical properties are described in detail both from an experimental and a modeling point of view.
6.2 The relationship between corrosion and hydrogen pickup
Already in the early studies, it was shown that hydride formation strongly correlates with corrosion rate [Citation2]. Several studies have shown that oxide layer thickness increased in regions of high hydride concentration, indicating that Zr-hydrides corrode faster than zirconium-alloy metal [Citation10]. It was demonstrated that once the TSS was reached, the corrosion started to accelerate, showing a post-transition corrosion rate as a function of hydrogen concentration [Citation127, Citation128, Citation137]. It was found that varies as a function of oxide thickness and the different fractions of hydrogen are picked up at different stages of oxide film growth [Citation11, Citation138].
The H pickup process follows the oxidation periodicity [Citation138, Citation139]; increases significantly from the initial pre-transition regime through subsequent transitions and drops just before the oxide transition [Citation31, Citation138, Citation139]. These variations repeat in the next transition regime following oxidation kinetic periodicity.
Couet et al. [Citation138] demonstrated, , that the hydrogen pickup mechanism is directly linked to the corrosion mechanism even though hydrogen pickup kinetics does not follow oxidation kinetics: the hydrogen pickup fraction increases from one transition regime to the next. Authors of [Citation138] hypothesized that the increase in is caused by changing one of the boundary conditions after the transition. The effect of hydrogen overpressure at the cathodic site has been identified as a possible cause of this hydrogen pickup fraction increase. It was also shown that the oxide resistivity followed a similar general evolution as
, indicating a possible correlation between oxide electronic conductivity, oxidation kinetics, and hydrogen pickup. It was fixed, that the corrosion temperature affected the onset of accelerated corrosion due to hydrogen, and this onset correlated with the TSS [Citation138].
Figure 18. Total hydrogen pickup fraction (determined experimentally and from the weight gain and hydrogen content fits) as a function of exposure time of (a) Zircaloy-4 sheet, (b) Zircaloy-4 tube, (c) ZIRLO® sheet, (d) ZIRLO® tube, (e) Zr–2.5Nb, (f) Zr–2.5Nb–0.5Cu. The transitions and the errors from the fits are also indicated, in [Citation138].
![Figure 18. Total hydrogen pickup fraction (determined experimentally and from the weight gain and hydrogen content fits) as a function of exposure time of (a) Zircaloy-4 sheet, (b) Zircaloy-4 tube, (c) ZIRLO® sheet, (d) ZIRLO® tube, (e) Zr–2.5Nb, (f) Zr–2.5Nb–0.5Cu. The transitions and the errors from the fits are also indicated, in [Citation138].](/cms/asset/33900fd0-1fed-48d7-bb3f-cbf469dc4d79/tnst_a_2127954_f0018_oc.jpg)
The experiment results of Wei et al. [Citation125] demonstrated that the corrosion kinetics accelerates in the presence of a hydride-rich rim, although the acceleration factor decreases with decreasing Sn content. Ni has been linked by B. Ensor et al. [Citation131] to higher hydrogen pickup, and if hydrogen concentration accelerates corrosion, then a higher pickup would lead to a faster acceleration of the corrosion rate.
B. Ensor et al. [Citation131] and T. Kim [Citation93] fixed the increase in corrosion rate caused by the high dissolved hydrogen level in the water. It was seen that the post-transition corrosion rate was increased with increasing hydrogen concentration. The corrosion acceleration is related to the precipitation of hydrides in the metal [Citation131], as the onset of the increased corrosion rate correlates with hydrogen concentration above the TSS of the alloy.
In the report of Romero et al. [Citation140], it is shown that thermal treatments, which increase the size of second-phase particles in Zircaloy-2 and Zircaloy-4 and induce phase transformations in ZIRLO and Zr-Nb, accelerate the onset of the oxidation transition, increase the oxidation rate after transition, and change the subsequent evolution of the hydrogen pickup fraction.
6.3 Impact of hydrides on the oxide layer properties
Mechanical response to the formation of hydrides has also been found to be a cause of the accelerated corrosion rate [Citation6, Citation130, Citation137, Citation141–143]: the formation of hydrides near the metal–oxide interface changes the local mechanical properties of the metal in that location. If the metal becomes harder and is less able to accommodate oxide growth stresses, critical stress at the metal–oxide interface could be reached sooner, leading to an earlier, local kinetic transition in the oxide. Microstructure observations of oxides grown on artificially formed hydride-rich rims have shown high levels of porosity [Citation130, Citation137, Citation141, Citation142]. It has been suggested that the complex stress field at the metal/oxide interface will result in the fracture of the brittle hydrides, disturbing the coherency at the metal/oxide interface and therefore reducing the integrity of the inner barrier oxide layer. Alternatively, because the volume expansion during hydride formation (17%) reduces the overall Pilling-Bedworth ratio from 1.56 to 1.3 (i.e. for the oxidation of zirconium hydride), it has been suggested that this could result in lower compressive stresses in the oxide and it is difficult to see how these two arguments can be affected by alloy chemistry.
Kim et al. in [Citation93] and [Citation143] observed for different Zr alloys that tetragonal zirconia developed during oxide growth and a fraction of the phase in the pre-hydrided alloy was consistently lower than that of an un-hydrided one. They suggested that the dissolved hydrogen may cause meta-stabilization of the tetragonal phase oxide grown at the metal–oxide interface.
The oxide morphology changes have been demonstrated in [Citation131]: as the hydrogen content increases, the oxide grains become more equiaxed and less columnar, , and the spacing between transition layers decreases with increasing hydrogen content. Additionally, locations of advanced oxide growth were correlated with locations of hydrides in the metal, : the oxide advance is greater where the hydrides appear to intersect the metal–oxide interface. This leads to a more uneven metal–oxide interface and a longer length of the metal–oxide interface leading to a higher corrosion rate due to the increased area where the oxygen can access the metal.
Figure 19. SEM images of 0.4 mm thick Zircaloy-4 coupons corroded for 397 days, (8.7 mm, 370 wt ppm hydrogen, left) and 1804 days, (71.6 mm, 2560 wt ppm hydrogen, right) in an autoclave at 360°C. Locations of transition cracks are shown with blue (dashed) arrows, modified from [Citation131].
![Figure 19. SEM images of 0.4 mm thick Zircaloy-4 coupons corroded for 397 days, (8.7 mm, 370 wt ppm hydrogen, left) and 1804 days, (71.6 mm, 2560 wt ppm hydrogen, right) in an autoclave at 360°C. Locations of transition cracks are shown with blue (dashed) arrows, modified from [Citation131].](/cms/asset/4eb6f3ea-85fe-4ecc-9636-8097b2021990/tnst_a_2127954_f0019_oc.jpg)
Figure 20. SEM micrograph of Zircaloy-4 sample 100,039 corroded for 1804 days at 360°C (71.6 mm). The oxide growth was accelerated along locations of hydrides, as highlighted with red arrows, from [Citation131].
![Figure 20. SEM micrograph of Zircaloy-4 sample 100,039 corroded for 1804 days at 360°C (71.6 mm). The oxide growth was accelerated along locations of hydrides, as highlighted with red arrows, from [Citation131].](/cms/asset/1672ed5e-132c-4a62-831e-e815736aae3b/tnst_a_2127954_f0020_oc.jpg)
Iglesias et al. [Citation36] noticed that hydrogen absorption can result in a sharp increase in the heat release with the dissolution of only 10% of the corrosion-product hydrogen in cladding without an oxide scale, i.e. while not as exothermic as oxidative heating. The cumulative heat release will decrease with a reduction in the solubility of hydrogen in zircaloy as the temperature increases. De Menibus and co-authors [Citation126] indicated that greater hydrogen contributions lead to increased material dilatation during heating. That was shown that hydrogen-induced expansion is anisotropic, it is important to consider the material texture to quantitatively reproduce the material expansion.
It is useful to mention [Citation132] that the terminal solid solubility for hydride precipitation during cooling is different from the terminal solid solubility for hydride dissolution during heating. The difference between these two concentrations is important for many applications.
7 Irradiation
Neutrons, γ, β, and α radiation, and possibly fission fragments influence the alloys, the oxide film, the CRUD, and the coolant. Their collective and interactive effects change the materials, corrosion mechanisms, and corrosion rates. Besides the obvious problems in experimental observations of irradiation, the impossibility to distinguish the radiation, thermal and chemical effects near control rods makes it further difficult to investigate the radiation impact on the alloy oxidation. These various processes need to be accounted for interpretation of radiation effects on zirconium alloy cladding. Phenomena that are more pronounced under heat transfer (e.g. crud deposition and concentration of species such as lithium) may impose a significant influence that is not readily reproducible on unheated surfaces [Citation127].
Although the thermo-hydraulic and chemistry influence much more the corrosion, irradiation affects the oxidation process by acting through [Citation6, Citation40, Citation59, Citation127, Citation128, Citation144]:
the change of the microstructure and element structure of the oxide film;
the phase stability of secondary phase particles and their concentrations;
dissolution/modification of SPPs;
the local electrical properties which affect the local electrochemistry;
the water chemistry (radiolysis);
the hydride rim formation.
Normally, it is a combination of various impacts initiated with irradiations and strongly influenced by operating conditions so, that this integrated effect can accelerate or weaken the corrosion.
There is little or no information in the open literature about the analysis of irradiation influence on the molecular level. It is still not clear how fast neutrons can increase diffusion rates in the protective oxide resulting in direct enhancement of the corrosion rates [Citation59], or how in-reactor irradiation changes the oxidation mechanism resulting in enhanced corrosion: through increased oxygen vacancies (or oxygen anions), or the resulting electric field across the oxide [Citation59], or due to changes in oxide morphology.
7.1 Irradiation influences the microstructure of the oxide film
The primary contributors to enhanced corrosion due to irradiation are the modifications of the metal microstructure, morphological and structural changes of the SPPs, concentrations of dissolved impurities, and characteristics of inclusions [Citation145–147]. Adamson and Rudling [Citation148] have given a historical review of the effect of irradiation on the microstructure of Zircaloy. The effect of irradiation is shown in [Citation148, Citation149], where the weight gain as a function of the SPPs at high fluence is given.
Motta and co-workers [Citation11, Citation145, Citation147] have studied the effect of electron and ion irradiation on Zr(Fe, Cr)2 and Zr2(Fe, Ni) particles in Zircaloy-2, and Zircaloy-4. They fixed the amorphization and decreased the sizes of particles. The amorphous layer was depleted of iron, and the thickness was proportional to the irradiation dose. It is considered that amorphization takes place because of a change in the stoichiometric composition of particles due to the escape of iron atoms. At higher temperatures, a redistribution of alloying elements resulted in the formation of precipitates in the matrix and at the grain boundaries. Sn-rich precipitates are also formed at these temperatures, due to radiation-enhanced diffusion [Citation150].
Figure 21. Effect of SPP size on corrosion of Zircaloy-2 cladding, from [Citation149].
![Figure 21. Effect of SPP size on corrosion of Zircaloy-2 cladding, from [Citation149].](/cms/asset/a37afd50-f7ea-4270-bebf-86326a8e2409/tnst_a_2127954_f0021_b.gif)
Etoh and Shimada [Citation151] also concluded that the rate-controlling step during the amorphization of Zr(Fe, Cr)2 particles is the diffusion rate of iron. The dissolution of Zr2(Fe, Ni) during neutron irradiation has passed without amorphization [Citation151].
Nishino et al. [Citation116] performed laboratory corrosion tests for three different zirconium alloys, which were based on Zircaloy-2, both with and without 60Co γ-ray irradiation, at 288°C oxygenated pure water, for 100 days. Corrosion weight gains of the alloy which had the lowest nodular corrosion resistance were lower for the irradiated condition than the non-irradiated condition. On the other hand, the alloys which had higher nodular corrosion resistances showed almost the same weight gains for both conditions. Differences in weight gain with and without irradiation were attributed to the dissolution of the oxide film in the high-temperature water. Dissolution tests of single-crystal yttria-doped ZrO2 indicated that 30–40% larger amounts dissolved into water under the γ-ray irradiation. Low-angle incident X-ray diffraction analysis showed that the tetragonal-ZrO2 fraction in the oxide film was lower with irradiation than without it, especially for the near-surface area.
The protective surface of the oxide layer ZrO2 is destroyed by the bombardment of ions. The point defects and dislocation loops induced by ion irradiation provide a fast diffusion channel to the oxygen atom from the surface to the matrix [Citation144] enhancing the oxidation of ions irradiated Zr-4 alloys with increased irradiation dose. On the other hand, the bombardment of ions changes the properties of the surface of the oxide layer influencing its electrochemical properties [Citation144].
7.2 Interaction between irradiation and alloy chemistry
Markelov et al. [Citation152] have performed autoclave corrosion tests of pre-irradiated and unirradiated fuel cladding samples made of Zr-Nb and Zr-Nb-Sn-Fe system alloys with different compositions. The composition of solid solutions from the matrix of alloys was significantly changed under irradiation compared to the unirradiated state. It was found that irradiation reduces corrosion of Zr-Nb alloys containing from 1% to 2.5% of Nb, and this effect becomes more apparent with increased content of Nb in the alloy. For Zr-Nb-Sn-Fe alloys, irradiation leads to more severe corrosion due to the increased content of Sn. The increased content of Nb and reduced content of Fe in the alloys of this system contributed to higher corrosion resistance. A structural indicator [Citation152] of alloy corrosion reduction under irradiation was the formation of nanometric niobium-rich radiation-induced precipitates (RIPs) due to the Nb exit from a matrix under irradiation. This process was significantly faster in Zr-Nb alloys. In Zr-Nb-Sn-Fe alloys, the presence of Sn in the initial solid solution of an alloy and the transition of Fe atoms thereto under irradiation retain the output of Nb atoms from the matrix to form RIPs.
To study changes in the oxidation state of tin under irradiation, specimens of Zr-0.76Fe-1.6Sn were corroded under similar conditions in an autoclave and a nuclear reactor by Filippov et al. [Citation153]. Corrosion rates were found to be significantly accelerated following irradiation compared to those in the autoclave. The kinetics of both the change in tin oxidation state and the corrosion rate in the reactor was greatly accelerated, pointing to the effect of irradiation driving tin into solution more quickly. Unlike that Yang et al. [Citation154] showed that the combined action of high residual compressive stress, supersaturated alloying elements, selective purification effect, and abundant structure defects in the modified layer helped improve the corrosion resistance, . They studied the combined action of high residual compressive stress, supersaturated alloying elements, selective purification effect, and abundant structure defects on corrosion resistance. Observations of microstructures showed that after high-current pulsed electron beam (HCPEB) surface irradiation, the surface layer of the sample was melted with the formation of gradually refined grains, martensites, and their internal twin substructures inside of the layer. Meanwhile, SPPs were dissolved into the matrix within the modified layer, and alloying elements Fe, Cr, and Nb were supersaturated in a Zr-based solid solution. In comparison to the initial sample, the pulsed sample was far more compact, micro-defects were hard to be observed, had fewer micro-pores on the inner surface and outer oxide layer, less quantity of cracks in the oxide film, and noticeably thinner oxide film. It was shown [Citation154] that all the oxide films, initial pulsed irradiation samples after 1 day and 30 days exposure, were composed of monoclinic ZrO2 and tetragonal ZrO2 phases. The authors of [Citation154] supposed that the compressive stress caused by HCPEB irradiation could cancel out a large portion of the tensile stress generated in the outermost layer of the Zr matrix and consequently decrease the transformation of metastable t-ZrO2 phase into stable m-ZrO2 phase. A higher concentration of alloying elements in the oxide film can not only restrain the nucleation of ZrO2 grains but also stabilize the t-ZrO2 phase.
Figure 22. Fracture surface SEM morphologies of the oxide films grown on initial (a-low magnification, c-high magnification) and 25-pulsed irradiation (b-low magnification, d-high magnification) samples after 30 days exposure, from [Citation154].
![Figure 22. Fracture surface SEM morphologies of the oxide films grown on initial (a-low magnification, c-high magnification) and 25-pulsed irradiation (b-low magnification, d-high magnification) samples after 30 days exposure, from [Citation154].](/cms/asset/257b208b-6a80-4713-ae1c-0af1b721780c/tnst_a_2127954_f0022_b.gif)
8 Conclusions and outlook
The multi-dimensional, multi-scale, multi-phase, and multi-parametric corrosion process of Zr alloys requires an in-depth understanding of each process and their interaction for the development of a model with a suitable validity range to simulate reliably such a process. Although having been the subject of numerous studies, the relationships between the physicochemical, electrochemical, and crystallographic processes driving the Zr corrosion still do not have fundamental analytical descriptions. Most of these investigations are empirical, ‘problem-oriented,’ constrained by the operation conditions studied, and sometimes even contradictive. The reason for that lies in the:
complexity of corrosion processes in general and the specific operating conditions of the investigated process;
very high specialization in these investigations focusing on only one dominant phenomenon in most of the studies;
mostly empirical investigations of the oxidation kinetics strongly linked to the studied conditions;
limited number of comprehensive studies of
the combined effects of different factors,
effects that influence the cyclic oxidation kinetics besides the stress accumulation in the oxide,
the fundamental molecular processes in both the oxide (electrochemistry) and the oxidizing medium (radiochemistry),
the influence of specific temperature gradients during an operation inside the cladding on the physicochemical properties of oxide and hydrogen distribution in the cladding;
large uncertainty in the thermochemical properties of species in chemical systems and reaction rates;
the huge amount of collected data related to the Zr corrosion which is of different quality are distributed unevenly in the parameter space and are sometimes inconsistent, and thus unsuited for simple analyses.
Recent progress in large-scale, multi-physics simulation tools, experiment techniques, the generation of large amounts of data, and the development of modern machine learning techniques opens up new possibilities for data-intense analyses and scientific investigations can lead to remarkable progress in computational simulations of corrosion processes, their detection, prevention, and mitigation. In connection with that, the principal phenomena and factors influencing ZrO2 film formation/destruction can be divided into five clusters of the possible neural network of the Zr corrosion deep learning approach, see also
Chemical composition of alloy and alloy manufacturing
Fundamental electrochemical processes
Chemistry of coolant water
Hydrogen pickup
Irradiation
The integrated effect of alloy composition, SPPs, impurities, and irregularities in the crystal structure, fabrication, and thermo-mechanical treatment of the alloy significantly affects the corrosion behavior of Zr alloys. That defines properties of the non- or protective layer through the oxide layer microstructure, the preferential crystallite orientations, and a stress accommodation; the migration of oxygen atoms, ions, and free electrons through the metal and oxide barrier layer; the reaction rates on the metal/oxide and oxide/environment boundaries, in the oxide layer; cyclic kinetics of metal oxidation, i.e. number and size of periodic layers of ZrO2 layer, transition points and rate of oxidation within a cycle; possible places for hydrogen accumulation.
Electrochemistry, being a fundamental part of corrosion science, which integrates electrochemical and quanta-physical processes, is unfortunately much less investigated (or presented in the open literature). This holds in particular for the anodic polarization curves of Zr alloys, which characterize the corrosion modes: start of corrosion, active corrosion, passivation, pitting, and the trans-passive regime depending on the alloy composition, temperature, medium nature, and pH. Future work is certainly required to investigate types of the Zr alloy polarization curves under different conditions, and the galvanic series of metals and alloys to predict/prevent potential galvanic corrosion situations and galvanic effects in the oxide layer provoked by SPPs.
Water chemistry affects all materials in contact with cooling water and is at the same time affected by the materials themselves. So, coolant chemistry is a multi-component multiphase reaction system defined by the interaction of five main chemical systems: the reactor coolant with different chemical additives such as H3BO3 and LiOH, KOH, hydrogen, oxygen, halogenides, sulfates, chromates, nitrates, etc.; water radiolysis; the possible escaped corrosion products and their hydroxides and aqueous ions; dissolution of Zr oxides and hydroxides; deposition of particulate and colloidal metal oxides, dissolved and hydrated metal ions (CRUD). Water chemistry parameters are studied mostly empirically with a focus on the development of water content control tools. The major aims of theoretical efforts should be directed at the development of
models for water radiolysis;
models to describe the impact of coolant pH and models for Zr hydrolysis;
models for the dissolution of zirconium oxide into the water, and dissolution of corrosion products into the primary coolant;
models for CRUD formation.
Technical guidance for the standardization of chemistry control in the nuclear industry internationally, like those available for the fossil-fueled power industry, would be very useful.
The hydrogen pickup leads to the accumulation of hydrogen, the formation of brittle hydrides and zirconium hydride platelets, and corrosion acceleration. The main hydrogen sources are hydrogen initially contained in the zirconium cladding and fuel pellets, cathodic generated H on the oxide layer/water boundary, the radiolysis dissociation of the coolant water, and the H2 gas dissolved in the primary cooling circuit. The hydrogen pickup affects the local mechanical properties of the metal and the oxide layer; accelerates oxide growth into the metal in the locations of precipitated hydrides near the metal–oxide interface; may cause meta-stabilization of the tetragonal phase oxide grown at the metal–oxide interface; changes the oxide morphology, equiaxed oxide grains versus the columnar grain structure; leads to a more uneven metal–oxide interface increasing the surface area between the metal and the oxide, and observed corrosion rates; increases the thermal expansion; accelerates the periodicity of the transitions in the cyclic Zr oxidation; increases the heat release with the dissolution of the corrosion-product. The correlation between oxidation kinetics and hydrogen pickup suggests a common corrosion mechanism such that any description of the hydrogen pickup should fit into a general corrosion mechanism.
The major effects of irradiation on the oxidation process are the change of the microstructure and element structure of the oxide film; the phase stability/dissolution/modification of secondary phase particles and their concentrations; the local electrical properties that affect the local electrochemistry; the water chemistry (radiolysis); the hydride rim formation. The fundamental mechanism of the irradiation impact on the corrosion process is not fully understood and thus an analytical description is still missing.
All listed factors are strongly influenced by the individual operating conditions: temperature, pressure, pH, hydrodynamic conditions and thermal gradients, duration, power of irradiation, etc.
Acknowledgments
This work was supported by the Federal Ministry for the Environment, Nature Conservation, Nuclear Safety and Consumer Protection (BMUV) of Germany (Bundesministerium für Umwelt, Naturschutz, nukleare Sicherheit und Verbraucherschutz (BMUV) Deutschlands), Contract-Nr.: 808875-0076.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Whitmarsh C. Review of Zirkolay-2 and zirkolay-4 properties relevant to N.S. Savannah reactor design. (TN): OAK Ridger National Laboratory, OAK Ridg; 1962.
- Parfenov B, Gerasimov V, Venediktova G. Corrosion of Zr and its alloys (in Russia). Moscow: Atomizdat; 1967. p. 246.
- Lemaignan C, Motta AT. Chapter 7: zirconium alloy in nuclear applications. In: Cahn RW, Haasen P, Kramer EJ, editors. Materials science and technology. Vol. 10B. Weinheim (Germany): VCH Verlagsgesellschaft mbH; 1994. p. 1–51.
- Schweitzer P. Metallic materials: physical, mechanical, and corrosion properties. Bosa Roca, United States: Taylor & Francis Inc; 2003.
- Adamson R, Rudling P. Manufacturing of Zr alloy materials. Molnlycke, Sweden: ANT International; 2004. (ZIRAT6/IZNA1 special topics report).
- Cox B. Some thoughts on the mechanisms of in-reactor corrosion of zirconium alloys. J Nucl Mater. 2005;336(2–3):331–368.
- Lemaignan C. Corrosion of zirconium alloy components in light water reactors. In: Cramer SD, Covino BS Jr., editors. Corrosion: environments and Industries. Vol. 13C. Metals Park (OH): ASM International; 2006. p. 415–420.
- I. A. E. AGENCY. Corrosion of zirconium alloys in nuclear power plants. Vienna: IAEA; 1993. (IAEA-TECDOC-684).
- Allen TR, Konings RJM, Motta AT, et al. Corrosion of zirconium alloys. In: Comprehensive nuclear materials. Vol. 5. Oxford (UK): Elsevier Ltd; 2012. p. 49–68.
- Rudling P, Adamson R, Garzarolli F, et al. Zr alloy corrosion and hydrogen pickup. Mölnlycke (Sweden): ANT International; 2013.
- Motta A, Couet A, Comstock RJ. Corrosion of zirconium alloys used for nuclear fuel cladding. Ann Rev Mater. 2015;45(1):311–343.
- Kim Y-S, Jeong Y-H, Jang J-N. Stress measurements during thin film zirconium oxide growth. J Nucl Mater. 2011;412(2):217–220.
- Adamson RB, Cox B, Garzarolli F, et al. Corrosion of zirconium alloys. Molnlycke, Sweden:ANT International; 2002/2003. (ZIRAT7 special topics report).
- Hillner E. Corrosion of zirconium base alloys — an overview. In: Zirconium in the Nuclear Industry: 3rd Int’l Symposium; West Conshohocken, PA; 1977.
- Motta A, Capolungo L, Chen L-Q, et al. Hydrogen in zirconium alloys: a review. J Nucl Mater. 2019;518:440–460.
- Stansbury EE, Buchanan RA. Fundamentals of electrochemical corrosion, materials park. Ohio: ASM International; 2000.
- Adamson R, Garzarolli F, Cox B, et al. Corrosion mechanisms in zirconium alloys. Skultuna, Sweden: ANT International; 2007. (IZNA7 special topic report).
- Haurais F. In: Evaluate the contribution of the fuel cladding oxidation process on the hydrogen production from the reflooding during a potential severe accident in a nuclear reactor. Saint-Aubin, France: Université Paris-Saclay; 2016.
- Reidy RF, Simkovich G. Electrical conductivity and point defect behaviour in ceria-stabilized zirconia. Solid State Ion. 1993;62(1–2):85–97.
- French RH, Glass SJ, Ohuchi FS, et al. Experimental and theoretical determination of the electronic-structure and optical properties of 3 phases of ZrO2. Phys Rev B. 1994;49(8):5133–5141.
- Valot C, Ciosmak D, Lallemant M. Spatiotemporal dynamics in the oxidation of groups IV-V metals: study of zirconium. Solid State Ion. 1997;101-103:769–774.
- Srinivasan R, Rice L, Davis B. Critical particle size and phase transformation in zirconia: transmission electron microscopy and X-ray diffraction studies. J Am Ceram Soc. 1990;73(11):3528–3530.
- Gosmain L, Valot C, Ciosmak D, et al. Study of stress effects in the oxidation of Zircaloy-4. Solid State Ion. 2001;141-142:633–640.
- Chao C-Y, Lin LF, Macdonald D. A point defect model for anodic passive films. I. Film growth kinetics. J Electrochem Soc. 1981;128(6):1187–1194.
- Yoo H-I, Koo B-J, Honh J-O, et al. A working hypothesis on oxidation kinetics of zircaloy. J Nucl Mater. 2001;299(3):235–241.
- Schefold J, Lincot D, Ambard A, et al. The cyclic nature of corrosion of Zr and Zr-Sn in high-temperature water (633 K): a long-term in situ impedance spectroscopic study. J Electrochem Soc. 2003;150(10):B451–B461.
- Betova I, Bojinov M, Laitinen T, et al. The transpassive dissolution mechanism of highly alloyed stainless steels I. Experimental results and modelling procedure. Corros Sci. 2002;44(12):2675–2697.
- Krausova A, Macak J, Sajdl P, et al. In-situ electrochemical study of Zr1nb alloy corrosion in high temperature Li+ containing water. J Nucl Mater. 2015;467(Part_1):302–310.
- Barberis P, Frichet A. Characterization of zircaloy-4 oxide layers by impedance spectroscopy. J Nucl Mater. 1999;273(2):182–191.
- Renciukova V, Macak J, Sajdl P, et al. Corrosion of zirconium alloys demonstrated by using impedance spectroscopy. J Nucl Mater. 2018;510:312–321.
- Tupin M, Pijolat M, Valdivieso F, et al. Differences in reactivity of oxide growth during the oxidation of zircaloy-4 in wate rvapour before and after the kinetic transition. J Nucl Mater. 2003;317(2–3):130–144.
- Peters H. Improved characterisation of aqueous corrosion kinetics of zircaloy-4. Philadelphia (PA): American Society for Testing and Materials; 1984.
- Wei J, Frankel P, Polatidis E, et al. The effect of Sn on autoclave corrosion performance and corrosion mechanisms in Zr–Sn–Nb alloys. Acta Materialia. 2013;61(11):4200–4214.
- Lyapin A, Jeurgens L, Mittemeijer E. Effect of temperature on the initial, thermal oxidation of zirconium. Acta Materialia. 2005;53(10):2925–2935.
- Simic N. Anodic Films on Cadmium and Binary Zirconium Alloys [PhD Thesis]. Göteborg (Sweden): Göteborg University; 2000.
- Iglesias F, Lewis B, Desgranges C. Clad-coolant chemical interaction (NEA-NSC-R--2015-5) State-of-the-Art Report on Multi-scale Modelling of Nuclear Fuels. In: Nuclear Energy Agency of the OECD (NEA): 2015;pp. 108–122.
- Motta A. Waterside corrosion in zirconium alloys. JOM. 2011;63(8):59–63.
- Nicholls RJ, Ni N, Lozano-Perez S, et al. Crystal structure of the ZrO phase at zirconium/zirconium oxide interfaces. Adv Eng Mater. 2015;17(2):211–215.
- De Gabory B, Motta A, Wang K. Transmission electron microscopy characterization of Zircaloy-4 and ZIRLO™ oxide layers. J Nucl Mater. 2015;456:272–280.
- Motta AT, Couet A, Comstock RJ. Corrosion of zirconium alloys used for nuclear fuel cladding. Ann Rev Mater. 2015;45(1):311–343.
- Yilmazbayhan A, Motta A, Comstock R, et al. Structure of zirconium alloy oxides formed in pure water studied with synchrotron radiation and optical microscopy: relation to corrosion rate. J Nucl Mater. 2004;324(1):6–22.
- Preuss M, Frankel P, Lozano-Perez S. Towards a mechanistic understanding of corrosion mechanisms in zirconium alloys. In Zirconium in the nuclear industry: 16th Int’l Symposium; Chengdu, China; 2010.
- Yilmazbayhan A, Breval E, Motta A, et al. Transmission electron microscopy examination of layers formed on Zr alloys. J Nucl Mater. 2006;349(3):265–281.
- Adamson RB, Cox B, Garzarolli F, et al. ZIRAT11/IZNA6 annual report. Molnlycke, Sweden: ANT International; 2006/2007.
- Wang P. Corrosion Behaviour of Zirconium Alloys in High Temperature Aqueous Environment By Electrochemical Impedance Spectroscopy [PhD Thesis]. University of Manchester: Corrosion & Protection Centre; 2011.
- Tang C, Stuebr M, Seifert H, et al. Protective coatings on zirconium-based alloys as accident-tolerant fuel (ATF) claddings. Corros Rev. 2017;35(3):141–165.
- Agency IAE. Waterside corrosion of zirconium alloys in nuclear power plants. IAEA-TECDOC-996. Vienna: IAEA; 1998.
- Cox B, Ungurelu M, Wong YM, et al. Mechanisms of LiOH degradation and H3BO3 repair of ZrO2. In: Zirconium in the nuclear industry: 11th Int’l Symposium; Garmisch-Partenkirchen (Germany); 1996.
- Polatidis E, Frankela P, Weia J, et al. Residual stresses and tetragonal phase fraction characterisation of corrosion tested Zircaloy-4 using energy dispersive synchrotron X-ray diffraction. J Nucl Mater. 2013;432(1–3):102–112.
- Lustman B, Kerze F, Straumanis ME. The metallurgy of zirconium. J Electrochem Soc. 1957;104(12):254C.
- Zima G. A review of the properties of zircaloy-2. 1959. (Technical Report No HW-60908).
- Banerjee S, Kamath HS. Materials for nuclear reactor. 2005. (Technical Report).
- Hong H, Kim S, Lee K. Influence of dilute silicon addition on the oxidation resistance and tensile properties of modified Zircaloy-4. J Nucl Mat. 2002;304(1):8–14.
- Nikulina A, Shishow S, Cox B, et al. Manufacturing of Zr-Nb Alloys. ANT International, Skultuna (Sweden), 2006; ZIRAT-11 Special Topic Report.
- Mondal K, Chatterjee UK, Murty BS. Oxidation behavior of multicomponent Zr-based amorphous alloys. J Alloys Compd. 2007;433(1–2):162–170.
- Park J, Kim H, Jeong Y, et al. Crystal structure and grain size of Zr oxide characterized by synchrotron radiation microdiffraction. J Nucl Mater. 2004;335(3):433–442.
- Jeong Y, Kim HG, Kim DJ. Influence of Nb concentration in the -matrix on the corrosion behavior of Zr-xNb binary alloys. J Nucl Mater. 2003;323(1):72–80.
- Jeong Y, Lee K, Kim H. Correlation between microstructure and corrosion behavior of Zr-Nb binary alloy. J Nucl Mater. 2002;302(1):9–19.
- Motta AT, Yilmazbayhan A, Comstock RJ, et al. Microstructure and growth mechanism of oxide layers formed on Zr alloys studied with micro-Beam synchrotron radiation. J ASTM Int. 2005;2(5):205–232.
- Broy Y, Garzarolli F, Seibold A, et al. Influence of transition elements Fe, Cr, and V on long-time corrosion in PWRs. In: Zirconium in the Nuclear Industry: 12th Int’l Symposium, ASTM STP 1354; West Conshohocken, PA; 2000.
- Adamson R, Cox B, Rudling P, et al. The annual review of zirconium alloy technology for 2000. Molnlycke, Sweden: ANT International; 2000. (ZIRAT5/IZNA1 Annual Report).
- Barberis P, Ahlberg E, Simic N, et al. Role of the second-phase particles in zirconium binary alloys. In: Zirconium in the Nuclear Industry: 13th Int’l Symposium; West Conshohocken, PA; 2002.
- Huang K, Logé R. A review of dynamic recrystallization phenomena in metallic materials. Mater Des. 2016;111:548–574.
- Kass S. The development of the zircaloys. In: Symposium on Corrosion of Zirconium Alloys; West Conshohocken, PA; 1964.
- Cox B. Mechanisms of zirconium alloy corrosion in nuclear reactors. J Corros Sci Eng. 2003;6:14.
- Bojinov M, Karastoyanov V, Kinnunen P, et al. Influence of water chemistry on the corrosion mechanism of a zirconium–niobium alloy in simulated light water reactor coolant conditions. Corros Sci. 2010;52(1):54–67.
- Cheng B, Krüger RM, Adamson RB. Corrosion behaviour of irradiated zircaloy. Proceedings of the 10. international symposium on zirconium in the nuclear industry. 21-24 Jun. Baltimore, MD (United States).1993.
- Cheng B, Adamson R. Mechanistic studies of zircaloy nodular corrosion. In: Zirconium in the Nuclear Industry: 7th Int’l Symposium; Philadelphia; 1987.
- Brossmann U, Würschum R, Södervall U, et al. Oxygen diffusion in ultrafine grained monoclinic ZrO2. J App Phys. 1999;85(11):7646.
- Gong W, Zhang H, Wu C, et al. The role of alloying elements in the initiation of nanoscale porosity in oxide films formed on zirconium alloys. Corros Sci. 2013;77:391–396.
- Garzarolli F, Seidel H, Tricot R, et al. Oxide growth mechanism on zirconium alloys. ASTM Spec Tech. 1991;1132:395–415.
- Garner A, Hu J, Harte A, et al. The effect of Sn concentration on oxide texture and microstructure formation in zirconium alloys. Acta Mater. 2015;99:259–272.
- Anghel C, Hultquist G, Limbäck M. Influence of Pt, Fe/Ni/Cr-containing intermetallics and deuterium on the oxidation of Zr-based materials. J Nucl Mater. 2005;340(2–3):271–283.
- Oskarsson M. Study on the mechanisms for corrosion and hydriding of zircaloy [Doctoral Thesis]. Stockholm (Sweden): Royal Institute of Technology, Division of Mechanical Metallurgy, Department of Materials Science and Engineering; 2000.
- Goossens A, Vazquez M, Macdonald D. The nature of electronic states in anodic zirconium oxide films part 1: the potential distribution. Electrochim Acta. 1996;41(1):35–45.
- Damaskin B, Petrii O, Tsirlina G. Electrochemistry. Moscow: Khimiya; 2001. p. 624.
- Roberge P. Corrosion engineering. New York: McGraw-Hill Education; 2008.
- Sharma SK, ed. Green corrosion chemistry and engineering: opportunities and challenges. Weinheim (Germany): Wiley-VCH Verlag GmbH & Co. KGaA; 2012.
- Sato N. Basics of corrosion chemistry. In: Sharma SK, editor. Green corrosion chemistry and engineering: opportunities and challenges. Weinheim (Germany): Wiley‐VCH Verlag GmbH & Co. KGaA; 2011. p. 1–32.
- Chen X, Bai X, Deng P, et al. Potential-pH diagram of Zr-H2O system at the increased temperatures. Rare Metal Mat Eng. 2004;33(7):710–713.
- Mamun A, Schennac R, Parga J, et al. Passive film breakdown during anodic oxidation of zirconium in pH 8 buffer containing chloride and sulfate. Electrochim Acta. 2001;46(22):3343–3350.
- Abdel Rahim M, Abdel Rahman A, Khalil M. Anion incorporation and its effect on the dielectric constant and growth rate of zirconium oxides. J Appl Electrochem. 1996;26(10):1037–1043.
- Bojinov M, Fabricius G, Laitinen T, et al. Transpassivity mechanism of iron-chromium-molybdenum alloys studied by AC impedance, DC resistance and RRDE measurements. Electrochim Acta. 1999;44(24):4331–4343.
- Betova I, Bojinov M, Laitinen T, et al. The transpassive dissolution mechanism of highly alloyed stainless steels II. Effect of pH and solution anion on the kinetics. Corros Sci. 2002;44(12):2699–2723.
- Song G. Transpassivation of Fe-Cr-Ni stainless steels. Corros Sci. 2005;47(8):1953–1987.
- Fattah-Alhosseini A, Saatchi A, Golozar M, et al. The transpassive dissolution mechanism of 316L stainless steel. Electrochim Acta. 2009;54(13):3645–3650.
- Ramasubramanian N. Shadow corrosion. J Nucl Mater. 2004;328(2–3):249–252.
- Mahmood S, Cantonwine P, Lin Y, et al. Shadow corrosion-induced bow of zircaloy-2 channels. In: XVI-th Intern. Symposium on Zirconium in the Nuclear Industry; Chengdu, China; 2010.
- Chatelain A, Andersson B, Ballinger RG, et al. Enhanced corrosion of zirconium-base alloys in proximity to other metals: the ‘shadow’ effect. In: ANS Int. Topical Meeting on LWR Fuel Performance; Park City, Utah, USA; 2000.
- Melehovets A, Pyshin I. Galvanic corrosion of zirconium alloys in water coolant. Izvestiya vuzov Yadernaya Energetika. 2020;2(2):52–63.
- Lister D, Uchida S. Determining water chemistry: 50th anniversary invited review. J Nucl Sci Technol. 2015;52(4):451–466.
- Cox B, Wu C. Dissolution of zirconium oxide film in 300°C LiOH. J Nucl Mater. 1993;199(3):272–284.
- Kim T, Choi KJ, Yoo SC, et al. Influence of dissolved hydrogen on the early stage corrosion behavior of zirconium alloys in simulated light water reactor coolant conditions. Corros Sci. 2018;131:235–244.
- Pêcheur D, Godlewski J, Peybernès J, et al. Contribution to the understanding of the effect of the water chemistry on the oxidation kinetics of zircaloy-4 cladding. In: Zirconium in the nuclear industry: 12th Int’l Symposium; West Conshohocken, PA; 2000.
- E. P. R. Institute. Pressurized water reactor primary water chemistry guidelines: revision 6. Palo Alto (CA): EPRI; 2007.
- I. A. E. Agency. High temperature on-line monitoring of waterchemistry and corrosion control in water cooled powerreactors. Vienna; 2002. (IAEA TECDOC-1303).
- Duriez C, Drouan D, Pouzadoux G. Reaction in air and in nitrogen of pre-oxidised Zircaloy-4 and M5™ claddings. J Nucl Mater. 2013;441(1–3):84–95.
- Coolant technology of water cooled reactors.Volume 1: Chemistry of primary coolantin water cooled reactors. Vienna, Austria: Int Atomic Energy Agency. 1992. IAEA -TECDOC-667.
- Mesmer RE, Baes CF, Sweeton FH. Acidity measurements at elevated temperatures. VI. Boric acid equilibriums. Inorg Chem. 1972;11(3):537–543.
- Macdonald DD, Urquidi-Macdonald M, Mahaffy JH, et al. Electrochemistry of water-cooled nuclear, Final technical progress report. University Park (PA): Pennsylvania State University; 2006.
- Liu W, Zhou B, Li Q, et al. Detrimental role of LiOH on the oxide film formed on Zircaloy-4. Corros Sci. 2005;47(7):1855–1860.
- Une K, Sakamoto K, Aomi M, et al. Hydrogen absorption mechanism of zirconium alloys based on characterization of oxide layer. In: Zirconium in the nuclear industry: 16th Int’l Symposium; Chengdu, China; 2010.
- Pêcheur D, Godlewski J, Billot P, et al. Microstructure of oxide films formed during the waterside corrosion of the zircaloy-4 cladding in lithiated environment. In: Zirconium in the nuclear industry: 11th Int’l Symposium; West Conshohocken, PA; 1996.
- Une K, Sakamoto K, Aomi M, et al. Hydrogen absorption mechanism of zirconium alloys based on characterization of oxide layer. J ASTM Int. 2011;8(5):1–21.
- Betova I, Bojinov M, Saario T. Start-up and shut-down water chemistries in pressurized water reactors. Espoo, Finland; 2012. (VTT Research Report, No VTT-R-00699-12).
- Steinbrück M, Schaffer S. High-temperature oxidation of zircaloy-4 in oxygen–nitrogen mixtures. Oxid Met. 2016;85(3–4):245–262.
- Bojinov M, Cai W, Kinnunen P, et al. Kinetic parameters of the oxidation of zirconium alloys in simulated WWER water – effect of KOH content. J Nucl Mater. 2008;378(1):45–54.
- E. P. R. Institute. Materials reliability program: effect of zinc addition on mitigation of primary water stress corrosion cracking of alloy 600 (MRP-78). Palo Alto (CA): EPRI; 2002.
- Betova I, Bojinov M, Kinnunen P, et al. Zn injection in pressurized water reactors – laboratory tests, field experience and modelling. Finland; 2011. (Research Report No. VTT-R-05511-11).
- Nordmann F, Odar S, Venz H, et al. ANT International chemistry update and best practices. Paper 9.02. In: Nuclear Plant Chemistry Conference, NPC 2010; Quebec, Canada; 2010.
- Byers A. Zinc and CRUD, how they interact. In: EPRI Fuel Reliability Program WG1 Meeting; Las Vegas, USA; 2005.
- Elliot A. Rate constants and g-values for the simulation of the radiolysis of light water over the range 0-300 deg C. 1994 Oct. (AECL Report No. 11073).
- Rishel D, Eklund K, Kammenzind B. In-situ EIS measurements of irradiated zircaloy-4 post-transition kinetic behavior. In: Zirconium in the nuclear industry: Proc. of the 15th Int’l Symposium; Sunriver Oregon, USA; 2007.
- Ai J, Chen Y, Urquidi-Macdonald M, et al. Electrochemical impedance spectroscopic study of passive zirconium, I. High-temperature, deaerated aqueous solutions. J Electrochem Soc. 2007;154(1):C43–C51.
- Ai J, Chen Y, Urquidi-Macdonald M, et al. Electrochemical impedance spectroscopic study of passive zirconium, II. High-temperature, hydrogenated aqueous solutions. J Electrochem Soc. 2008;154(1):C52–C59.
- Nishino Y, Endo M, Ibe E, et al. Formation and dissolution of oxide film on zirconium alloys in 288°C pure water under γ-ray irradiation. J Nucl Mater. 1997;248:292–298.
- Christensen H. Remodeling of the oxidant species during radiolysis of high-temperature water in a pressurized water reactor. Nucl Tech. 1995;109(3):373–382.
- Bertuch A, Pang J, Macdonald D. The argument for low hydrogen and lithium operation in PWR primary circuits. In: 7th international symposium on environmental degradation of materials in nuclear power systems: Water reactors; Breckenridge, CO; 1995.
- Macdonald DD, Urquidi-Macdonald M. Theory of steady-state passive films. J Electrochem Soc. 1990;137(8):2395–2402.
- Mirza NM, Rafique M, J HM, et al. Computer simulation of corrosion product activity in primary coolants of a typical PWR under flow rate transients and linearly accelerating corrosion. Ann Nucl Energy. 2003;30(7):831–851.
- Duro L, Grivé M, Cera E, et al. Update of a thermodynamic database for radionuclides to assist solubility limits calculation for PA. Stockholm (Sweden): Svensk Kärnbränslehantering AB, Swedish Nuclear Fuel and Waste Management Co; 2005.
- Shikina N, Medvedkina O, Popova E, et al. Experimental study of the ZrO2 solubility in water solutions of perchloric acid at 150°С. Vestn Otd nauk Zemle. 2011;3(NZ6099):1–7.
- Curti E, Degueldre C. Solubility and hydrolysis of Zr oxides: a review and supplemental data. Radiochim Acta. 2002;90(9–11):801–804.
- Kritskiy A. Solubility of zirconium and chromium corrosion products in aqueous solutions at 298-623 K, St. Petersburg, Russian Federation [Ph.D. Thesis]. St. Petersburg Technological Institute; 1992.
- Wei J, Frankel P, Blat M, et al. Autoclave study of zirconium alloys with and without hydride rim. Corros Eng Sci Technol. 2012;47(7):516–528.
- De Menibus A, Guilbert T, Auzoux Q, et al. Hydrogen contribution to the thermal expansion of hydrided Zircaloy-4 cladding tubes. J Nucl Mater. 2013;440(1–3):169–177.
- Pêcheur D, Lefebvre F, Motta A, et al. Oxidation of intermetallic precipitates in Zircaloy-4: impact of irradiation. In: Zirconium in the nuclear industry: 10th Int’l Symposium; Philadelphia, PA; 1994.
- Agency IAE. Review of fuel failures in water cooled reactors. In: Nucl. Energy Ser. NF-T-2.1. Vienna, Austria; 2010.
- Cox B, Y.-m W. Hydrogen uptake micro-mechanism for Zr alloys. J Nucl Mater. 1999;270(1–2):134–146.
- Blat M, Legras L, Noel D, et al. Contribution to a better understanding of the detrimental role of hydrogen on the corrosion rate of Zircaloy-4 cladding materials, in: zirconium in the Nuclear Industry: STP 1. Proceedings of the 12th International Symposium, ASTM STP 1354; 1998; Toronto, Canada; West Conshohocken, Pa., USA: ASTM International.
- Ensor B, Lucente A, Frederick M, et al. The role of hydrogen in zirconium alloy corrosion. J Nucl Mater. 2017;496:301–312.
- Courty O, Motta AT, Hales J. Modeling and simulation of hydrogen behavior in Zircaloy-4 fuel cladding. J Nucl Mater. 2014;452(1–3):311–320.
- Une K, Ishimoto S. Dissolution and precipitation behavior of hydrides in Zircaloy-2 and high Fe Zircaloy. J Nucl Mater. 2003;322(1):66–72.
- Bertolino G, Meyer G, Perez IJ. In situ crack growth observation and fracture toughness measurement of hydrogen charged Zircaloy-4. J Nucl Mater. 2003;322(1):57–65.
- Zielinski A, Sobieszczyk S. Hydrogen-enhanced degradation and oxide effects in zirconium alloys for nuclear applications. Int J Hydrogen Energy. 2011;36(14):8619–8629.
- Dauma R, Chu Y, Motta A. Identification and quantification of hydride phases in Zircaloy-4 cladding using synchrotron X-ray diffraction. J Nucl Mater. 2009;392(3):453–463.
- Wang X, Zheng M-J, Szlufarska I, et al. Continuum model for hydrogen pickup in zirconium alloys of LWR fuel cladding. Int J Appl Phys. 2017;121(13):135101.
- Couet A, Motta A, Comstock R. Hydrogen pickup measurements in zirconium alloys: relation to oxidation kinetics. J Nucl Mater. 2014;451(1–3):1–13.
- Harada M, Wakamatsu R. The effect of hydrogen on the transition behavior of the corrosion rate of zirconium alloys. In: Zirconium in the Nuclear Industry: 15th Int’l Symposium; West Conshohocken, PA; 2008.
- Romero J, Partezana J, Comstock R, et al. Evolution of hydrogen pickup fraction with oxidation rate on zirconium alloys. Top Fuel Reactor Fuel Performance, Zurich, Switzerland. 2015;476–482.
- Platt P, Polatidis E, Frankel P, et al. A study into stress relaxation in oxides formed on zirconium alloys. J Nucl Mater. 2015;456:415–425.
- Wang X, Khafizov M, Szlufarska I. Effect of surface strain on oxygen adsorption on Zr (0 0 0 1) surface. Int J Appl Phys. 2014;445(1–3):1–6.
- Kim Y, Jeong Y, Son S. A study on the effects of dissolved hydrogen on zirconium alloys corrosion. J Nucl Mater. 2014;444(1–3):349–355.
- Yan C, Wang R, Wang Y, et al. Effects of ion irradiation on microstructure and properties of zirconium alloys—A review. Nucl Eng Technol. 2015;47(3):323–331.
- Motta AT, Lemaignan C, Olander DR. Segregation of tin in zircaloy-2 under proton irradiation. In: 15th Int’l Symposium on the Effects of Irradiation on Materials; Nashville, TN; 1992.
- Garzarolli F, Goll W, Seibold A, et al. Effect of in-PWR irradiation on size, structure, and composition of intermetallic precipitates of Zr alloys Zircon. Proceedings of Eleventh international symposium: Zirconium in the nuclear industry. American Society for Testing and Materials.(1996)
- Pêcheur D, Lefebvre F, Motta AT, et al. Effect of irradiation on the precipitate stability in Zr alloys. J Nucl Mater. 1993;205:445–451.
- Adamson RB, Rudling P. Properties of zirconium alloys and their applications in light water reactors (LWRs). In: K.L. Murty,editor. Materials ageing and degradation in light water reactors. Cambridge (UK): Woodhead Publishing; 2013. p. 151–245.
- Garzarolli F, Schumann R, Steinberg E. Corrosion optimized zircaloy forBoiling Water Reactor (BWR) fuel elements. In: Tenth Int’l Symposium, Zirconium in the Nuclear Industry, ASTM STP 1245; Philadelphia; 1994.
- Griffiths M, Gilbert RW, Cheadle BA. Formation, growth and decomposition of precipitates in zircaloy-2 and −4, AECL-8852. Atomic Energy of Canada, Chalk River Nuclear Labs: Chalk River, Ontario; 1985.
- Etoh Y, Shimada SJ. Neutron irradiation effects on intermetallic precipitates in Zircaloy as a function of fluence. J Nucl Mater. 1993;200(1):59–69.
- Markelov V, Novikov V, Shevyakov A, et al. Preliminary irradiation effect on corrosion resistance of zirconium alloys.In: Zirconium in the Nuclear Industry: 18th Int’l Symposium; West Conshohocken, PA; 2018.
- Filippov V, Bateev A, Lauer Y, et al. The influence of reactor irradiation on the oxidation state of tin in Zr-0.76Fe-1.6Sn. Hyperfine Interact. 2018;239(1):21.
- Yang S, Guo Z, Zhao L. Surface microstructures and high-temperature high-pressure corrosion behavior of N18 zirconium alloy induced by high current pulsed electron beam irradiation. Appl Surf Sci. 2019;484:453–460.