Abstract
We developed protocols to analyse the cell cycle of Heterosigma akashiwo in the natural phytoplankton assemblage using tyramide signal amplification-fluorescence in situ hybridisation (TSA-FISH) and flow cytometry. We determined the optimum probe and formamide concentrations for the species-specific TSA-FISH probe to be 0.5 ng µL−1 and 40%, respectively. The probe did not hybridise to non-target phytoplankton including Chattonella sp., which is closely related to Heterosigma and has high sequence similarity to it. We could successfully identify G1- and G2-phase cells and the diel cell cycle of H. akashiwo not only in the laboratory but also in the field after TSA-FISH. These results demonstrate that TSA-FISH for H. akashiwo in natural coastal waters could serve as a powerful tool for understanding their bloom mechanism using flow cytometry.
Introduction
Heterosigma akashiwo causes red tides in coastal environments and has been recognised on a global scale as having a negative impact on fisheries (Hard et al. Citation2000; Khan et al. Citation1997). In particular, this species caused massive kills of yellowtail and red sea bream in Japan and Chinook salmon in New Zealand and Canada (Chang et al. Citation1990; Honjo Citation1993). H. akashiwo blooms in New Zealand are long lasting in late spring (Rhodes et al. Citation1993). In Asia, blooms of this species occur rapidly and disappear dramatically within three days in coastal waters from spring to autumn (Han & Furuya Citation2000). Rapid bloom formation could be explained by the ability of H. akashiwo to multiply more than four times a day in outdoor tanks and in laboratory experiments (Honjo & Tabata Citation1985). However, the growth rate of H. akashiwo is not well-documented in natural environments (Honjo Citation1993).
To obtain a better understanding of the bloom development mechanism, accurate estimates of species-specific in situ growth rates are needed for the migration and bloom mechanisms of H. akashiwo red tide. The bottle incubation shares a common shortcoming in which precise estimation of the species-specific growth rate is impossible because various phytoplankton assemblages are mixed in with the natural organisms. Even though it could evaluate the species-specific growth rate, Rivkin and Seliger's (Citation1981) method, which is single-cell 14C assay, is not easy to conduct and requires incubation. However, application of the aforementioned methods mentioned may not be feasible in open ocean systems (Campbell & Carpenter Citation1986). Therefore, a different approach is potential growth rate (McDuff & Chisholm Citation1982). This method is based on cell cycle analysis of the fraction of cells in a key stage (Campbell & Carpenter Citation1986; Chang & Carpenter Citation1991; Pan & Cembella Citation1997; Reguera et al. Citation2003). The estimation of in situ growth rate based on the cell cycle analysis has been used particularly for prokaryote picophytoplankton such as genera Prochlorococcus and Synechococcus in the ocean (Binder & Chisholm Citation1995; Binder Citation2000; Bruyant et al. Citation2005; Chisholm Citation1995; Jacquet, Partensky, Lennon et al. Citation2001; Jacquet, Partensky, Marie Citation2001). When Prochlorococcus and Synechococcus were analysed with flow cytometry, they were easily discriminated from the other phytoplankton assemblages because of their small size and autofluorescent properties. Still, detection of a certain harmful algal bloom (HAB) species using flow cytometry is difficult because of overlaps in size and similar fluorescence intensities with non-target species. Therefore, cell cycle analysis for eukaryotic phytoplankton in natural environments has been limited, although it has been investigated for several algal cultures in the laboratory (Pan & Cembella Citation1997; Peperzak et al. Citation1997).
Several techniques were developed to identify and enumerate H. akashiwo, including sandwich hybridisation (Tyrrell et al. Citation2001; Tyrrell et al. Citation2002) and polymerase chain reaction (PCR)-based methods (Asai et al. Citation2003; Akase et al. Citation2004). Recently, fluorescence in situ hybridisation (FISH) has been developed for rRNA and rDNA, a rapid and cost-effective method for species identification (Chen et al. Citation2008). However, this detection technique still has a problem; the signal is not sufficient to detect the cells using flow cytometry. Therefore, we applied a tyramide signal amplification (TSA)-FISH technique, which increases fluorescence intensity (Not et al. Citation2002) for sensitive and accurate identification and cell cycle analysis in natural environments. Furthermore, the development of a fixative was also needed to protect fragile H. akashiwo cells from the processes of the TSA-FISH and cell cycle analyses. Paraformaldehyde (PFA) is a well-known fixative, which penetrates rapidly into the cell, and sodium cacodylate has been frequently used as a buffer to preserve cell morphology in electron microscopy (Tiffany et al. Citation2001). Thus, we combined PFA and sodium cacodylate to form a rapidly effective fixative. For this study, a new specific probe was designed to detect H. akashiwo. As the probe concentration strongly influenced probe reactivity, an optimum probe concentration for the TSA-FISH was also determined, and we investigated the suitable formamide concentration for signal amplification. Finally, we confirmed the species specificity of the probe and the possibility of cell cycle analysis after TSA-FISH in the laboratory and in the natural environment.
Materials and methods
Sampling, cultured strains and cell fixation
For the probe specificity test, natural seawater samples were collected at Geoje Island, in the southern part of Korea, on 9 June 2006. For the cell cycle analysis, natural water samples were collected every 4 h at Shiwha Lake in the western part of Korea from 23 to 24 May 2007, during which time H. akashiwo had formed a bloom. All water samples were fixed immediately, and the samples were transported to the laboratory on ice and stored at −70 °C until hybridisation.
Table 1 Strains used in the present study and their probe reactivities for the HSIG 1451 probe.
The cultured strains used in this study are shown in . All of the strains were cultured with F/2 medium at 20 °C under 60–100 µE m−2 s−1 with a 12:12 L:D cycle. Cells of H. akashiwo were fixed with PFA at a final concentration of 0.1% (). The fixative contains sodium cacodylate (pH 7.4, 0.2 M, final concentration), sucrose (0.1 M, final concentration) and Pluronic F-68 cell surfactant (0.1%, final concentration).
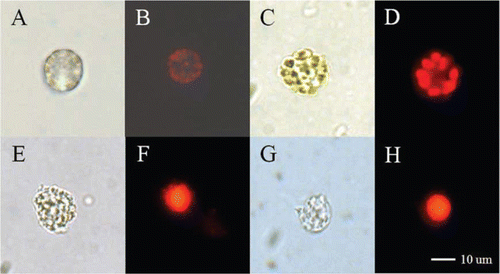
Figure 1 Light and fluorescence micrographs of Heterosigma akashiwo strain HYM06HA. A, Live cell. B, Chlorophyll in the live cell. C, Cell fixed with paraformaldehyde (PFA). D, Chlorophyll in the fixed cell. E, Cell following chlorophyll extraction with methanol after PFA fixation. F, Nucleus stained with propidium iodide (PI) after methanol treatment. G, Cell following chlorophyll extraction with ethanol after PFA fixation. H, Nucleus stained with PI after ethanol treatment.
Probe design
Oligonucleotide probe designs used in this study () are available from the literature, except for HSIG 1451, which was designed to target the species Heterosigma akashiwo (). The probe EUK 1209 was used as a positive control and probe NON 338 was used as a negative control. The probe sequence was determined with the probe design function of the ARB software (http://www.arb-home.de) using a database of rRNA gene sequences with more than 14,000 sequences. Before designing the probe, we added more than 60 sequences of raphidophyceae and dinophyceae. Probes were screened with BLAST searches (http://www.ncbi.nlm.nih.gov/BLAST/) to examine their specificities against a wide range of organisms. Melting temperatures (Tm) for the probe were checked following the method of Hugenholtz et al. (Citation2001) using the nearest neighbor method.
Table 2 Oligonucleotide probes used in the present study.
Table 3 Specificities of the HSIG1451 probe.
Fluorescence in situ hybridisation for flow cytometry
For in situ hybridisation, 35 mL of H. akashiwo culture was fixed for 1 h at room temperature in 50 mL conical tubes (Falcon, USA). The cell surfactant Pluronic F-68 (10% stock solution; Sigma-Aldrich) was added to samples at a final concentration of 0.1%. Cells were collected via centrifugation (4000 g for 3 min at 4 °C), and the pellet was re-suspended in c. 20 mL of phosphate-buffered saline (PBS; Sigma-Aldrich) solution at 1x and pH 7.4, containing 0.1% Pluronic F-68, stored at 4 °C. For cell dehydration, cells were collected using centrifugation (3600 g, 3 min) and dehydrated through the addition of c. 5 mL of 96% ethanol. We confirmed that ethanol treatment removed chlorophyll, which interferes with propidium iodide (PI; Sigma, USA) fluorescence (). Chlorophyll-free cells were either directly processed for in situ hybridisation or were stored at −30 °C for several weeks.
A portion of the samples was washed with 1 mL of distilled water (DW) amended with 0.1% of Pluronic F-68 in a 1.5 mL centrifuge tube. Centrifugation was performed with a 5417R bench-top centrifuge (Eppendorf, France) at 3600 g for 3 min. After removing the DW, 30 µL of hybridisation buffer (0.9M NaCl, 20 mM Tris-HCl [pH 7.4], 0.01% sodium dodecyl sulfate [Sigma, USA], 10% dextran sulfate [SDS, Sigma, USA], 2% blocking reagent [Sigma, USA], 40% formamide [Sigma, USA]) was added. To reduce cell aggregation, tubes were sonicated for 10 s (55 W, 35 kHz, UT-53N, Sharp, Japan). Oligonucleotide probes (0.6 µL) labelled with horseradish peroxidase (HRP, 25 ng DNA µL−1) were added and incubated at 35 °C for 2.5 h. Nonhybridised probes were washed for 1 min at 35 °C with 1.5 mL of prepared buffer (56 mM NaCl, 5 mM EDTA, 0.01% SDS, 20 mM Tris-HCl [pH 7.5], 0.1% Pluronic F-68). A second washing step using 1 mL of the same buffer was carried out for 30 min.
Cells equilibrated with 1 mL of PBS (pH 7.4) were amended with c. 7% of Tween20 and 0.1% of Pluronic F-68 for 15 min at room temperature. Two steps of centrifugation removed most of the buffer. Twenty microlitres of TSA mix (40% dextran sulfate mixed 1:1 with the amplification diluents of the TSA direct kit [NEN Life Science Products, USA]) were added 50:1 to fluorescein-tyramide in the TSA direct kit. Tubes were sonicated for 10 s and incubated for 30 min in the dark at room temperature. Unlabelled fluorochrome was removed using two subsequent rinses of 1 min and 20 min in 1 mL of the PBS buffer. Flow cytometric analysis was conducted immediately.
Fluorescence in situ hybridisation for epifluorescence microscopy
Some algal species, especially Chattonella, form cell aggregates during the centrifugation steps. Therefore, filters were used to examine probe specificity via epifluorescence microscopy according to the method of Pernthaler et al. (Citation2002). Samples, fixed as mentioned previously, were filtered on 1.2 µm polycarbonate membrane filters (47 mmΦ; Whatman, USA). Filters were dipped in 0.2% of low-gelling-point agarose (Sigma), dried on slide glasses at 35 °C and were subsequently dehydrated in 96% ethanol for 1 min and dried.
Filters were cut into eight sections, each of which was placed into a 0.2 mL microtube; 150 µL of the hybridisation buffer described previously and 1.5 µL of the HRP probe working solution (50 ng DNA µL−1) were pipetted onto the filter sections. A formamide concentration of 40% was used for the examination. Microtubes were incubated at 35 °C for at least 2 h. Filters were removed from the hybridisation mixture and incubated at 35 °C for 20 min in the pre-warmed washing buffer described previously.
To equilibrate the probe-delivered HRP, sections were incubated in PBS amended with 7% Tween20 for 15 min at room temperature. Sections were dabbed onto blotting paper and immediately transferred to the substrate mixture as described previously. Filter sections were incubated at room temperature in the dark for at least 30 min, placed onto blotting paper to remove excess tyramide-fluorescein, and washed twice for 1 min in PBS amended with 7% Tween20, DW, and 96% ethanol. After drying for 15 min, the filter sections were incubated in PI solution for 5 min, washed with DW, and then mounted onto a slide glass with non-fluorescent immersion oil for immediate observation.
Flow cytometry
Fluorescence intensities of the cells were analysed with a flow cytometer (Calibur, Becton Dickinson, USA) equipped with an air-cooled argon laser (488 nm, 150 mW). The fluorescences of FL1 (Green, 515–545 nm), FL3 (Red, >600 nm) and the forward scatter (FSC) and side scatter (SSC) were recorded; FL1, FSC, and SSC were recorded in a logarithmic mode, and FL3H (peak height of FL3), FL3W (peak width of FL3), and FL3A (peak area of FL3) were recorded in a linear mode to analyse the relative DNA quantities of the cells for cell cycle analysis. All parameters were normalised with 6 µm fluorescent beads (Polyscience, USA). Fluorescence-activated cell sorting flow solution (Becton Dickinson, USA) was used as a sheath fluid. Cell aggregates were eliminated by gating the cytogram within the linear range, and histograms of the relative DNA amounts were analysed with the Modfit program (Verity Software House, Inc., Palo Alto, CA, USA) to quantify the percentage of cells in each of the cell stages (G1, S, G2 + M). At least 10,000 cells were analysed.
Epifluorescence microscopy
Cells were also observed with an epifluorescence microscope (Axioplan, Zeiss, Germany) under blue excitation, equipped with a 100-W mercury lamp and a 40×objective lens. Microphotographs were taken with a charge-coupled device camera (Qincom fast, Qimaging, Canada).
Cell cycle analysis of H. akashiwo in the natural samples
We confirmed the possibility for cell cycle analysis when the natural samples were treated with TSA-FISH. Cells treated with RNase (final concentration 20 mg mL−1, Sigma, USA) were incubated for 30 min at 35 °C, then were stained with 20 µg mL−1 PI. Cell cycle analysis was performed on a flow cytometer following the procedure described previously.
Results
For the TSA-FISH protocol and/or cell cycle analysis of H. akashiwo, fixation is a fundamental and essential technique. Fixed cells of H. akashiwo had better conserved morphological characters compared with those of live cells and/or those having undergone other treatments (A, 1C, 1E, 1G). We also confirmed the difference between ethanol and methanol treatments for the dehydration and chlorophyll extraction steps; there was no chlorophyll interruption to the PIfluorescence of the stained nuclei (B, 1F, 1H).
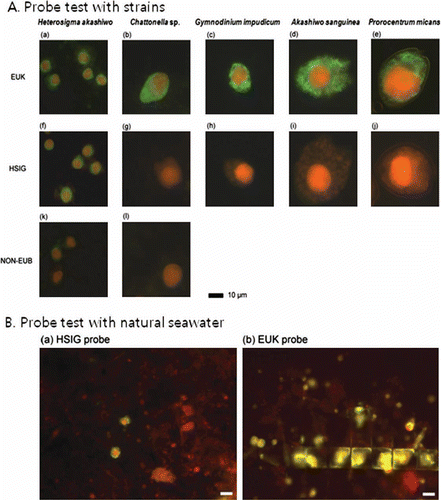
To establish the TSA-FISH protocol, we optimised the fluorescence intensities and formamide concentration. The EUK probe, which hybridises in eukaryotic organisms, was used as the positive control, while the antisense NON-EUB probe was used as the negative control. Fluorescence intensities of the NON-EUB probe in all tested concentrations were lower than those of the EUK probe (). However, when the fluorescence intensity of the NON-EUB probe exceeded 10 amplification units, it was difficult to distinguish between the positive and negative controls since the fluorescence for the NON-EUB probe overlapped with fluorescence for the EUK probe (data not shown). Consequently, discrimination of probe-positive and probe-negative cells was difficult when probe concentrations exceeded 2 ng µL−1. Therefore, we used a 0.5 ng µL−1 probe concentration for H. akashiwo cells. All formamide concentrations (30–70%) showed high fluorescence compared to that of the NON-EUB probe (). However, the fluorescence intensities of 50% and 60% formamide were greater than the fluorescence intensity of the EUK probe, indicating that when the formamide concentration was 50% or 60%, the fluorescence intensity was overestimated; therefore, we chose a 40% formamide level. To confirm the species specificity, we tested the cross-reactivities of three different H. akashiwo strains, Chattonella sp. and three common HAB causative species, Akashiwo sanguinea, Gymnodinium impudicum and Prorocentrum micans (). Also, the positive and negative controls were tested to define a reference for reactivity of a specific probe. When all species were treated with the positive control probe (EUK 1209), all cells exhibited green fluorescence (A[a]–4A[e]), while cells treated with the negative control probe (NON 338) exhibited no green fluorescence (A[k], 4A[l]). In contrast, in all strains tested for the HSIG 1451 probe, there was no nonspecific binding to other species (A). Specifically, we checked the specificity of the HSIG probe against the Chattonellastrain, which had the lowest number of mismatches (three) to the probe () in a formamide concentration series from 30% to 70%, and there was no nonspecific binding to Chattonella cells (A[g]). In the natural sample, H. akashiwo cells were successfully detected using this technique (B), and there was no nonspecific binding between the HSIG probe and other phytoplankton. TSA-FISH using the HSIG probe allowed for a clear and easy identification of H. akashiwo cells (B[a]), However, TSA-FISH using the EUK probe hybridised with various plankton in the natural assemblages (B[b]). All of these results illustrate that the H. akashiwo-specific probe designed in the present study makes possible the molecular identification of targeted species in natural assemblages.
Figure 2 Effect of probe concentration on fluorescent intensity (arbitrary units, AU) in Heterosigma akashiwo cells.
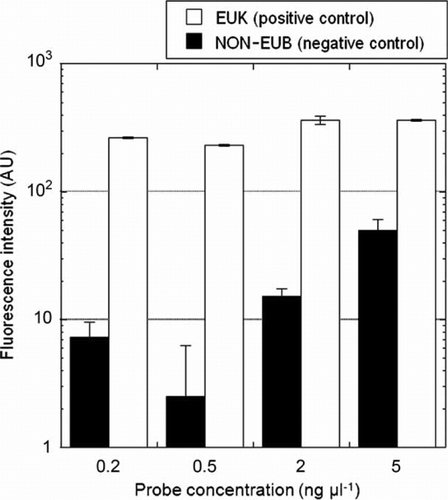
Figure 3 Effect of formamide concentration on fluorescent intensity (arbitrary units, AU) in Heterosigma akashiwo cells.
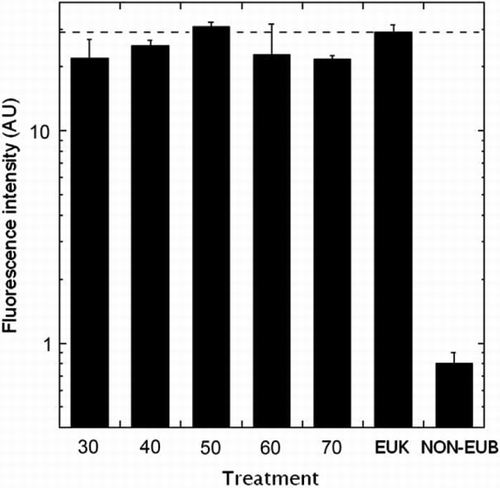
Figure 4 Epifluorescence micrographs. A, Algal cells after fluorescence in situ hybridisation using various probes. Panels a, f, and k: Heterosigma akashiwo, panels b, g and l: Chattonella sp., panels c and h: Gymnodinium impudicum, panels d and i: Akashiwo sanguine, panels e and j: Prorocentrum micans; upper panels (a, b, c, d, and e): cells hybridised with the EUK1209 probe, middle panels (f, g, h, I, and j): cells hybridised with the HSIG1451 probe, lower panels (k and l): cells hybridised with the NON338 probe. B, Natural seawater sample collected from Geoje Island. Panel a: cells hybridised with HSIG1451 probe, panel b: cells hybridised with EUK1209 probe.
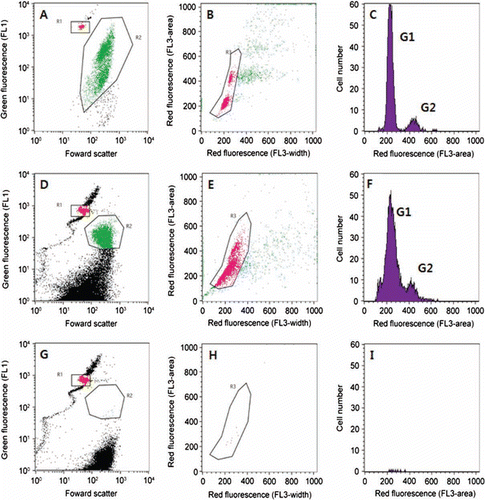
shows an example of the cell cycle analysis after TSA-FISH using a cultured H. akashiwo strain. After the TSA-FISH, cells of H. akashiwo had green fluorescence (A). Probe-positive cells were plotted by peak height (x-axis) and peak width (y-axis) of red fluorescence to remove the doublet cells (B). The DNA histogram of the singlet cells of H. akashiwo is gated in B and clearly shows peaks for G1- and G2-phase cells (C). The coefficient of variance for the G1-phase cells was sufficiently high for the analysis (8.28%). These results demonstrate that TSA-FISH can be used to analyse the amount of DNA in cells. Therefore, we successfully analysed the relative quantity of DNA for H. akashiwo cells in natural assemblages, although the coefficient of variance for G1-phase cells was 16% (). After the H. akashiwo cells were hybridised with the HSIG probe, target cells were detected using flow cytometry (D). We successfully identified G1- and G2-phase cells (F) through gating (E); however, H. akashiwo cells which were not hybridised to the NON-EUB probe were not detected (G–5I).
Figure 5 Flow cytometric signatures of Heterosigma akashiwo. A–C, Signatures of H. akashiwo strain after fluorescence in situ hybridization (FISH). D–I, Signatures of cells collected from Korean coastal waters after FISH. A, Detection of H. akashiwo cells after FISH; red dots indicate 6 m beads; green dots indicate H. akashiwo cells detected with green fluorescence after FISH. B, Discrimination of doublet and singlet cells of H. akashiwo; pink dots indicate singlet cells discriminated by peak area and peak width (length) of red fluorescence of detected cells (green dots) in panel A. C, DNA histograms of H. akashiwo gated in panel B (composed of singlet cells discriminated in panel B). D–F, Samples hybridised with HSIG1284 probe. G–I, Samples hybridised with NON338 probe.
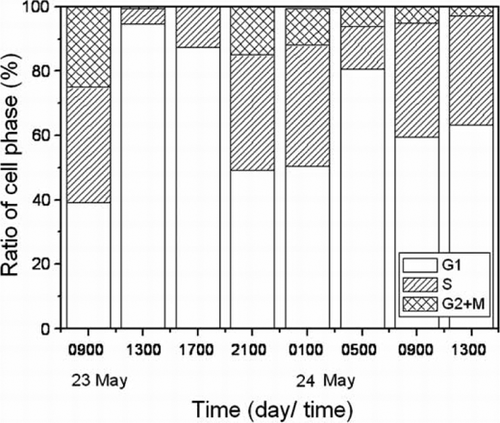
The diel cell cycle analysis of H. akashiwo in the natural assemblages is shown in . The percentage of G1-phase cells increased from 0900 h to 1300 h and the percentage of S-phase cells increased from 1700 h until 2100 h on 23 May. The percentage of G2 + M phase cells increased until 2100 h on 23 May. The percentage of G1-phase cells again increased at 0500 h on 24 May. To determine the timing of cell division, we focused on the G1-phase cells in this study. Based on these data, we observed that H. akashiwo synthesised DNA during the daytime, and we confirmed the application of this method for estimation of in situ growth rate.
Figure 6 Diel cell cycle analysis of Heterosigma akashiwo from Shiwha Lake after fluorescence in situ hybridisation.
Discussion
One of the important goals of phytoplankton ecology is prediction of harmful algal blooms in coastal environments. Bulk measurements and community-level studies mask the bloom-developing process of bloom-forming species (Collier Citation2000; Collier & Campbell Citation1999). Autecological approaches are more desirable for comparative and experimental investigations for the description of factors regulating algal blooms (Han et al. Citation1992; Han & Furuya Citation2000). It is useful to combine knowledge obtained in culture-based studies in the laboratory with that from single-cell-based studies in the natural ecosystem.
Formation of Heterosigma patches on the surfaces of coastal waters occurs quickly. One of the explanations is rapid growth; Honjo found that H. akashiwo cells divided three to four times per day in a laboratory experiment (Honjo Citation1993; Honjo & Tabata Citation1985). Interestingly, the division occurred during the 6 h dark periods. However, there is no report about a high growth rate in the natural ecosystem. Another explanation is aggregation of dispersed cells owing to a combination of swimming behaviour and physical processes (Bearon et al. Citation2006). It has been hypothesised that bloom formation of Heterosigma occurs at the freshwater interface because of heavy run-off from rivers. These findings are not sufficient to explain the reason for dense blooms of H. akashiwo, although it might be also one possible explanation for initial bloom formation. To understand the algal blooming mechanisms of H. akashiwo in the natural ecosystem, we proposed a new tool to estimate the species-specific growth rate and cell cycle using flow cytometry with the TSA-FISH method.
FISH has been used for the identification and enumeration of phytoplankton species in natural samples (Biegala et al. Citation2003; Lange et al. Citation1996; Miller & Scholin Citation1998; Parsons et al. Citation1999; Tyrrell et al. Citation2001). However, most DNA fluorochromes bind to both DNA and RNA; thus, it is desirable to remove all RNA prior to flow cytometric analysis. In this point of view, the standard FISH protocol using monolabelled oligonucleotides has no advantage for cell cycle analysis. In contrast, TSA-FISH does not utilise RNA after signal amplification, an important technical advantage of using TSA-FISH for cell cycle analysis in order to identify the target cell using flow cytometry.
In the application of TSA-FISH with H. akashiwo cells, it is necessary to explain some technical issues regarding this technique. We found that probe concentration is an important factor affecting the result of TSA-FISH (). When we used the 5 ng µL−1 concentrations generally used for un-amplified detection in FISH, the negative control appeared positive under the epifluorescence microscope (data not shown). Also, the fluorescence signal of the cells hybridised with the negative control probe was hardly discriminated from those hybridised with the positive control probe (data not shown). Yang et al. (Citation1999) recommended that the probe concentrations for TSA-FISH are 10 times lower than those recommended in many protocols for FISH with un-amplified detection. Indeed, many studies have used 0.5 ng µL−1 with TSA-FISH (Pernthaler et al. Citation2002). Therefore, the TSA-FISH technique has both a cost advantage and an effective sensitivity compared with those of FISH.
In a preliminary study of the cell cycle analysis with natural samples, problems were encountered. First, the TSA-FISH protocol based on HRP-labelled oligonucleotides permits the detection of small marine bacteria with low ribosomal content that are not or are only barely detectable with monolabelled probes. Secondly, the coefficient of variance for G1-phase cells was not sufficiently low (16%) to analyse the cell phase. However, we could easily determine the existence of G2-phase cells in the analysis. Thus, we could identify the timings of DNA replication and cell division of target species. Also, we successfully analysed the diel pattern of cell division of H. akashiwo in natural assemblages. Finally, PBS-buffered PFA has been frequently used for fixation in SEM or light microscopy. However, it was not suitable for fixation of H. akashiwo cells. This fixation sometimes causes white precipitation probably because of phosphate salt with divalent cations (Przysiezniak & Spencer Citation1989). Instead of PBS, the use of cacodylate prevents such precipitation. Owing to these methodological improvements, the cell morphology was well-preserved.
In conclusion, we developed a detection and cell cycle analysis technique for understanding the bloom development of H. akashiwo. This research served as a novel study to investigate the cell cycle of H. akashiwo in the field, despite its fragility. Also, the information obtained about the diel cell division pattern will provide important insights into the blooming mechanisms. Therefore, we believe this technique is a powerfultool for studying the blooming process of H.akashiwo in its natural environment.
Acknowledgements
We wish to thank two anonymous reviewers and Dr Judy Sutherland for comments on earlier versions that significantly improve the paper. This research was supported by the National Research Foundation of Korea (NRF-2008-314-C00319).
References
- Asai , R , Ootani , K , Nomura , Y , Nakamura , C , Ikebukuro , K , Arikawa , Y , Miyake , J and Karube , I . 2003 . PCR-based ribosomal DNA detection technique for microalga (Heterosigma carterae) causing red tide and its application to a biosensor using labeled probe . Marine Biotechnology , 5 : 417 – 423 .
- Akase , SI , Yoshikawa , T , Hayakawa , N , Maeda , H and Sakata , T . 2004 . Molecular identification of red tide-causing microalga Heterosigma akashiwo strains based on their chloroplast DNA sequences . Fisheries Science , 70 : 1043 – 1050 .
- Bearon , RN , Grunbaum , D and Cattolico , RA . 2006 . Effects of salinity structure on swimming behavior and harmful algal bloom formation in Heterosigma akashiwo, a toxic raphidophyte . Marine Ecology Progress Series , 306 : 153 – 163 .
- Biegala , IC , Not , F , Vaulot , D and Simon , N . 2003 . Quantitative assessment of picoeukaryotes in the natural environment by using taxon-specific oligonucleotide probes in association with tyramide signal amplification-fluorescence in situ hybridization and flow cytometry . Applied and Environmental Microbiology , 69 : 5519 – 5529 .
- Binder , B . 2000 . Cell cycle regulation and the timing of chromosome replication in a marine Synechococcus (cyanobacteria) during light- and nitrogen-limited growth . Journal of Phycology , 36 : 120 – 126 .
- Binder , BJ and Chisholm , SW . 1995 . Cell cycle regulation in marine Synechococcus sp. strains . Applied and Environmental Microbiology , 61 : 708 – 717 .
- Bruyant , F , Babin , M , Genty , B , Prasil , O , Behrenfeld , MJ , Claustre , H , Bricaud , A , Garczarek , L , Holtzendorff , J , Koblizek , M , Dousova , H and Partensky , F . 2005 . Diel variations in the photosynthetic parameters of Prochlorococcus strain PCC9511: combined effects of light and cell cycle . Limnology and Oceanography , 50 : 850 – 863 .
- Campbell , L and Carpenter , EJ . 1986 . Diel patterns of cell division in marine Synechococcus spp. (Cyanobacteria): use of the frequency of dividing cells technique to measure growth rate . Marine Ecology Progress Series , 32 : 139 – 148 .
- Chang , FH , Anderson , C and Bousted , NC . 1990 . First record of a Heterosigma (Raphidophyceae) bloom with associated mortality of cage-reared salmon in Big Glory Bay, New Zealand . New Zealand Journal of Marine and Freshwater Research , 24 : 461 – 469 .
- Chang , J and Carpenter , EJ . 1991 . Species-specific phytoplankton growth rates via diel DNA synthesis cycles. V. Application to natural populations in Long Island Sound . Marine Ecology Progress Series , 78 : 115 – 122 .
- Chen , GF , Wang , GC , Zhang , CY , Zhang , BY , Wang , XK and Zhou , BC . 2008 . Development of rRNA and rDNA-targeted probe for fluorescence in situ hybridization to detect Heterosigma akashiwo (Raphidophycea) . Journal of Experimental Marine Biology and Ecology , 355 : 66 – 75 .
- Chisholm , SW . 1995 . Cell cycle regulation in marine Synechococcus sp. strains . Applied and Environmental Microbiology , 61 : 708 – 717 .
- Collier , JL . 2000 . Flow cytometry and the single cell in phycology . Journal of Phycology , 36 : 628 – 644 .
- Collier , JL and Campbell , L . 1999 . Flow cytometry in molecular aquatic ecology . Hydrobiologia , 401 : 33 – 53 .
- Giovannoni , SJ , DeLong , EF , Olsen , GJ and Pace , NR . 1988 . Phylogenetic group-specific oligodeoxynucleotide probes for identification of single microbial cells . Journal of Bacteriology , 170 : 720 – 726 .
- Han , MS and Furuya , K . 2000 . Size and species-specific primary productivity and community structure of phytoplankton in Tokyo Bay . Journal of Plankton Research , 22 : 1221 – 1235 .
- Han , MS , Furuya , K and Nemoto , T . 1992 . Species-specific productivity of Skeletonema costatum (Bacillariophyceae) in the inner part of Tokyo Bay . Marine Ecology Progress Series , 79 : 267 – 273 .
- Hard , JJ , Connell , L , Hershberger , WK and Harrell , LW . 2000 . Genetic variation in mortality of chinook salmon during a bloom of the marine alga Heterosigma akashiwo . Journal of Fish Biology , 56 : 1387 – 1397 .
- Honjo , T . 1993 . “ Overview on bloom dynamics and physiological ecology of Heterosigma akashiwo ” . In Toxic phytoplankton blooms in the sea , Edited by: Smayda , TJ and Shimizu , Y . 33 – 41 . Elsevier : New York .
- Honjo , T and Tabata , K . 1985 . Growth dynamics of Olisthodiscus luteus in outdoor tanks with flowing coastal water and in small vessels . Limnology and Oceanography , 30 : 653 – 664 .
- Hugenholtz , P , Tyson , GW and Blackall , LL . 2001 . Design and evaluation of 16S rRNA-targeted oligonucleotide probes for fluorescence in situ hybridization . Methods in Molecular Biology , 176 : 29 – 41 .
- Jacquet , S , Partensky , F , Lennon , JF and Vaulot , D . 2001 . Diel patterns of growth and division in marine picoplankton in culture . Journal of Phycology , 37 : 357 – 369 .
- Jacquet , S , Partensky , F , Marie , D , Casotti , R and Vaulot , D . 2001 . Cell cycle regulation by light in Prochlorococcus strains . Applied Environmental Microbiology , 67 : 782 – 790 .
- Khan , S , Arakawa , O and Onoue , Y . 1997 . Neurotoxins in a toxic red tide of Heterosigma akashiwo (Raphidophyceae) in Kagoshima Bay, Japan . Aquaculture Research , 28 : 9 – 14 .
- Lange , M , Guillou , L , Vaulot , D , Simon , N , Amann , R , Ludwig , W and Medlin , LK . 1996 . Identification of the class Prymnesiophyceae and the genus Phaeocystis with ribsomal RNA-targeted nucleic acid probes 1 detected by flow cytometery . Journal of Phycology , 32 : 858 – 868 .
- McDuff , RE and Chisholm , SW . 1982 . The calculation of in situ growth rates of phytoplankton populations from fractions of cells undergoing mitosis: a clarification . Limnology and Oceanography , 27 : 783 – 788 .
- Miller , PE and Scholin , CA . 1998 . Identification and enumeration of cultured and wild Pseudo-nitzschia (Bacillariophyceae) using species-specific LSU rRNA-targeted fluorescent probes and filter-based whole cell hybridization . Journal of Phycology , 34 : 371 – 382 .
- Not , F , Simon , N , Biegala , IC and Vaulot , D . 2002 . Application of fluorescent in situ hybridization coupled with tyramide signal amplification (FISH-TSA) to assess eukaryotic picoplankton composition . Aquatic Microbial Ecology , 28 : 157 – 166 .
- Pan Y , Cembella AD 1997 . Flow cytometric determination of cell cycles and growth rates of Prorocentrum spp . In: Reguera B , Blanco J , Fernandez M , Wyatt T . Proceedings of the VIII international conference on Harmful Algae, Vigo, Spain, 25–29 June 1997 . Xanta de Galicia and Intergovermental Oceanographic Commission of UNESCO . Pp. 173 – 176 .
- Parsons , ML , Scholin , CA , Miller , PE , Doucette , GJ , Powell , CL , Fryxell , GA , Dortch , Q and Soniat , TM . 1999 . Pseudo-nitzchia species (Bacillariophyceae) in Louisiana coastal waters: molecular probe field trials, genetic variability, and domoic acid analysis . Journal of Phycology , 35 : 1368 – 1378 .
- Peperzak L , Sandee B , Jonker R , Legrand C 1997 . Measurement of Prorocentrum micans growth rate by flow cytometric analysis of the diel DNA cycle . In: Reguera B , Blanco J , Fernandez M , Wyatt T . Proceedings of the VIII international conference on Harmful Algae, Vigo, Spain, 25–29 June 1997 . Xanta de Galicia and Intergovermental Oceanographic Commission of UNESCO . Pp. 177 – 178 .
- Pernthaler , A , Pernthaler , J and Amann , R . 2002 . Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria . Applied and Environmental Microbiology , 68 : 3094 – 3101 .
- Przysiezniak , J and Spencer , AN . 1989 . Primary culture of identified neurons from a cnidarians . Journal of Experimental Biology , 142 : 97 – 113 .
- Reguera , B , Garces , E , Pazos , Y , Bravo , I , Ramilo , I and González-Gil , S . 2003 . Cell cycle patterns and estimates of in situ division rates of dinoflagellates of the genus Dinophysis by a postmitotic index . Marine Ecology Progress Series , 249 : 117 – 131 .
- Rhodes , LL , Haywood , AJ , Ballantine , WJ and MacKenzie , AL . 1993 . Algal blooms and climate anomalies in north-east New Zealand, August–December 1992 . New Zealand Journal of Marine and Freshwater Research , 27 : 419 – 430 .
- Rivkin , RB and Seliger , HH . 1981 . Liquid scintillation counting for C uptake of single algal cells isolated from natural samples . Limnology and Oceanography , 26 : 780 – 785 .
- Tiffany , M , Barlow , S , Matey , V and Hurlbert , S . 2001 . Chattonella marina (Raphidophyceae), a potentially toxic alga in the Salton Sea, California . Hydrobiology , 466 : 187 – 194 .
- Tyrrell , JV , Bergquist , PR , Bergquist , PL and Scholin , CA . 2001 . Detection and enumeration of Heterosigma akashiwo and Fibrocapsa japnoca (Raphidophyceae) using rRNA-targeted oligonucleotide probes . Phycologia , 40 : 457 – 467 .
- Tyrrell , JV , Connell , LB and Scholin , CA . 2002 . Monitoring for Heterosigma akashiwo using a sandwich hybridization assay . Harmful Algae , 1 : 205 – 214 .
- Wallner , G , Amann , R and Beisker , W . 1993 . Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms . Cytometry , 14 : 136 – 143 .
- Yang , H , Wanner , IB , Roper , SD and Chaudhari , N . 1999 . An optimized method for in situ hybridization with signal amplification that allows the detection of rare mRNAs . Journal of Histochemistry and Cytochemistry , 47 : 431 – 445 .