
Abstract
CYP2A13 enzyme is expressed in human extrahepatic tissues, while CYP2A6 is a hepatic enzyme. Reactions catalysed by CYP2A13 activate tobacco-specific nitrosamines and some other toxic xenobiotics in lungs.
To compare oxidation characteristics and substrate-enzyme active site interactions in CYP2A13 vs CYP2A6, we evaluated CYP2A13 mediated oxidation characteristics of 23 coumarin derivatives and modelled their interactions at the enzyme active site.
CYP2A13 did not oxidise six coumarin derivatives to corresponding fluorescent 7-hydroxycoumarins. The Km-values of the other coumarins varied 0.85–97 µM, Vmax-values of the oxidation reaction varied 0.25–60 min−1, and intrinsic clearance varied 26–6190 kL/min*mol CYP2A13). Km of 6-chloro-3-(3-hydroxyphenyl)-coumarin was 0.85 (0.55–1.15 95% confidence limit) µM and Vmax 0.25 (0.23–0.26) min−1, whereas Km of 6-hydroxy-3-(3-hydroxyphenyl)-coumarin was 10.9 (9.9–11.8) µM and Vmax 60 (58–63) min−1. Docking analyses demonstrated that 6-chloro or 6-methoxy and 3-(3-hydroxyphenyl) or 3-(4-trifluoromethylphenyl) substituents of coumarin increased affinity to CYP2A13, whereas 3-triazole or 3-(3-acetate phenyl) or 3-(4-acetate phenyl) substituents decreased it.
The active site of CYP2A13 accepts more diversified types of coumarin substrates than the hepatic CYP2A6 enzyme. New sensitive and convenient profluorescent CYP2A13 substrates were identified, such as 6-chloro-3-(3-hydroxyphenyl)-coumarin having high affinity and 6-hydroxy-3-(3-hydroxyphenyl)-coumarin with high intrinsic clearance.
Introduction
Humans are constantly exposed to a wide variety of foreign compounds (xenobiotics). The greatest exposure for xenobiotics takes place from the gastrointestinal tract, but significant exposure occurs also via lung. In the case of smokers or certain types of occupations exposure by inhalation can be the most significant exposure route when toxic effects are considered. Lipophilic xenobiotics are absorbed more than water-soluble and charged ones. Enzymatic phase 1 oxidation, reduction or hydrolysis and phase 2 conjugation reactions are the most important elimination pathway for the lipophilic xenobiotics. The liver is the major xenobiotic metabolising organ in the body, but metabolism is also important in tissues of entry, such as lung, intestine, and skin (Gonzalez et al. Citation2018; Parkinson et al. Citation2019).
The cytochrome P450s (CYP) are the most versatile metabolising enzymes for numerous xenobiotics and endogenous substrates. Of the 57 individual CYP enzymes in humans, approximately 10 forms in the families CYP1, CYP2 and CYP3 are expressed at high level in liver (Zanger and Schwab Citation2013). CYP enzymes are less abundant in extrahepatic tissues, making them quantitively less important for the overall metabolism of xenobiotics. Local metabolism, however, may lead to tissue-specific adverse responses, e.g. organ toxicities, allergies or cancer (Gundert-Remy et al. Citation2014).
The respiratory system consists of tissues that are ports of entry for inhaled chemicals. CYPs and other xenobiotic-metabolising enzymes are expressed at every level of the respiratory tract, starting from nasal epithelial, and ending in lung alveoli (Hukkanen et al. Citation2002; Oesch et al. Citation2019). Of the two functional enzymes in the human CYP2A subfamily, CYP2A6 is abundant in liver, whereas CYP2A13 is expressed in extrahepatic tissues. CYP2A13 is expressed especially in nasal mucosa and lung (Raunio and Rahnasto-Rilla Citation2012). CYP2A13 and CYP2A6 share 95.4% amino acid identity Fernandez-Salguero et al. Citation1995). In the respiratory system, several compounds are bioactivated from pro-toxic forms to ultimate toxic metabolites (Pelkonen and Raunio Citation1997; Anttila et al. Citation2011; Oesch et al. Citation2019). CYP2A13 mediated bioactivation plays a role in lung tumorigenesis. CYP2A13 activates many procarcinogens in tobacco, including the tobacco-specific N-nitrosamines 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and N9-nitrosonornicotine (NNN) (Su and Ding Citation2004; Jalas et al. Citation2005; Zhang et al. Citation2007), naphthalene, phenanthrene, biphenyl (Shimada et al. Citation2016; Li et al. Citation2017), and 5-hydroxymethylfurfural (Ji et al. Citation2018). Other notable examples of compounds metabolised by CYP2A13 are aflatoxins (He et al. Citation2006; Zhang et al. Citation2013), nicotine (Hukkanen et al. Citation2005; Murphy et al. Citation2005) and coumarin (Fukami et al. Citation2007).
Recently we designed new profluorescent coumarin substrates for human hepatic CYP enzymes These substrates are oxidised efficiently by CYP1A1, CYP1A2 and/or CYP1B1 and some of them also by CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4 (Juvonen et al. Citation2019; Juvonen et al. Citation2021 submitted). The primary aim of this study was to use these novel coumarin derivatives as tools to explore their CYP2A13 oxidation kinetics, and to compare the new compounds with the classical CYP substrates 7-ethoxy- and 7-pentoxyresorufin. The binding of the coumarins to the active site of human CYP2A13 was modelled in silico and compared to that of CYP2A6. Several novel coumarin derivatives proved to be oxidised more efficiently than coumarin by CYP2A13.
Materials and methods
Chemicals
MgCl2 were from Honeywell Riedel-de Haen (Bucharest, Romania). Ethanol (≥99.5%, Etax Aa) was from Altia (Helsinki, Finland). Water was deionised by MilliQ gradient A10. All chemicals were of the highest purity available from their commercial suppliers. Coumarin, 6-methylcoumarin (25), 7-hydroxycoumarin, 7-ethoxyresorufin, 7-pentoxyresorufin, resorufin, Tris-HCl, MnCl2, isocitric acid and isocitric acid dehydrogenase were purchased from Sigma-Aldrich (Steinheim, Germany), KCl from J.T. Baker, NADPH and NADP+ from Roche Diagnostics (Mannheim, Germany). 200 mL NADPH regenerating system contained 178.5 mg NADP+ (nicotinamide adenine dinucleotide phosphate), 645 mg isocitric acid, 340 mg KCl, 240 mg MgCl2, 0.32 mg MnCl2 and 15 U isocitric acid dehydrogenase.
Coumarin derivatives
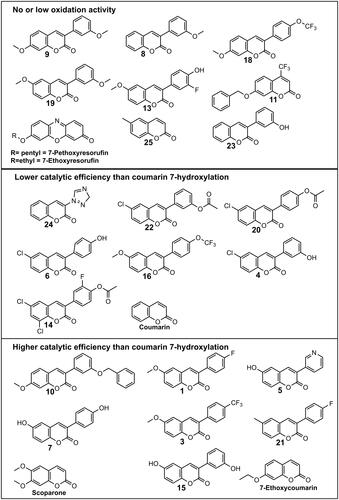
Synthesis and experimental data for compounds 3-(4-fluorophenyl)-6-methoxycoumarin (1); 3-(4-trifluoromethylphenyl)-6-methylcoumarin (2); 3-(4-trifluoromethylphenyl)-6-methoxycoumarin (3); 3-(3-hydroxyphenyl)-6-chlorocoumarin (4), 3-pyridyl-6-chlorocoumarin (5); 3-(4-hydroxyphenyl)-6-chlorocoumarin (6); 3-(4-hydroxyphenyl)-6-hydroxycoumarin (7); 3-(3-methoxyphenyl)coumarin (8); 3-(3-methoxyphenyl)-7-methoxycoumarin (9); 3-(3-benzyloxo)phenyl-7-methoxycoumarin (10); 4-trifluoromethyl-7-benzyloxocoumarin (11); 3-(3-fluoro-4-hydroxyphenyl)-6-methoxy-coumarin (13); 6,8-dichloro-3-(4-(3-fluoro)phenyl acetate)-coumarin (14); 3-(3-hydroxyphenyl)-6-hydroxy-coumarin (15); 7-methoxy-3-(4-(trifluoromethoxy)phenyl)-coumarin (16); 7-methoxy-3-(4-(trifluoromethoxy)phenyl)-coumarin (18); 6-methoxy-3-(3-methoxyphenyl)-coumarin (19); 3-(4-phenyl acetate)-6-chloro-coumarin (20); 3-(4-fluorophenyl)-6-methyl-coumarin (21); 3-(3-phenyl acetate)-6-chloro-coumarin (22); 3-(3-hydroxyphenyl)-coumarin (23); 3-(1H-1,2,4-triazol-1-yl)-coumarin (24) are described earlier (Niinivehmas et al. Citation2018; Rauhamäki et al. Citation2018; Juvonen et al. Citation2019; Citation2021). Structures of the molecules are shown in .
Figure 1. Structures of coumarin derivatives grouped by CYP2A13 mediated oxidation rate and efficiency. Numbers assigned to the derivatives are the same as in previous publications (Juvonen et al. Citation2019, Citation2021). Grouping was made according to intrinsic clearance (Vmax/Km) values presented in .
Biological material
Baculovirus-insect cell-expressed human CYP2A13 was purchased from BD Biosciences Discovery Labware (Woburn, MA, USA) and used according to the manufacturer’s instructions.
Oxidation assays
The kinetic assays were carried out in 100 µL volume containing 100 mM Tris-HCl buffer pH 7.4, 0–40 µM coumarin derivative or 0–10 µM 7-ethoxyresorufin, 1–25 nM recombinant CYP or 0–0.1 g/L microsomal protein and 20% NADPH regenerating system. Incubations took place at 37 °C in 96-multiwell plates; the fluorescence was measured with a Victor2 plate reader (PerkinElmer Life Sciences, Turku, Finland). The detailed conditions are described in the figures and tables. The reaction was started by adding NADPH and fluorescence was measured at 2-min intervals for 40 min using excitation 405 nm and emission 460 nm for the oxidation of coumarin derivatives and excitation 570 nm and emission 615 nm for 7-ethoxyresorufin or 7-pentoxyresorufin 7-O-dealkylations. Incubations without substrate, enzyme or NADPH were used as blank reactions. Resorufin was used as a standard and 7-hydroxycoumarin as the surrogate standards to calculate the amount of product formed. The linear phase of the reactions was used for calculations. When the oxidation of coumarin 7-hydroxylation or 7-hydroxylation of compound 15 was inhibited by 0–500 µM pilocarpine or 0–100 µM 7-methylcoumarin, the same incubation conditions and measurement setup as described above was used. One µL pilocarpine or 7-methylcoumarin was added from 100 mM phosphate pH 7.4 or 100% dimethyl sulfoxide stock solution, respectively. Non-inhibited sample contained 1% dimethyl sulfoxide, and negative control did not contain microsomes.
Multivariate analysis
For multivariate analysis of kinetic parameters, principal component analysis on mean centred and normalised kinetics data was done using SIMCA version 15.0.2, Umetrics (Simca multivariate data analysis software). Principal component analysis reveals underlying patterns in the data by reducing the dimensions of the data to uncorrelated variables (principal components). All obtained component scores were reduced by component scores from coumarin to force coumarin to the origin. This was done to ease interpretation, since all other compounds are derivates of coumarin.
Molecular docking
Ten compounds with the highest 7-hydroxylation efficiency were docked to the active site of CYP2A13 to predict their binding modes and to explain the observed differences in Km and Vmax values. The compounds were drawn with MAESTRO (ver. 12.5.139, Release 2020-3, Schrödinger, LLC) and prepared for docking with LIGPREP using the following settings: OPLS3e force field (Harder et al. Citation2016); protonation at pH 7.4 using EPIK (Shelley et al. Citation2007); maximum of 32 stereoisomers per ligand. LIGPREP preparation produced a single structure for each compound.
All compounds were docked to the five crystal structures of human CYP2A13 available in Protein Data Bank (PDB) (Berman et al. Citation2000): 2P85, 3T3S, 4EJG, 4EJH and 4EJI (Yano et al. Citation2005; Smith et al. Citation2007; DeVore et al. Citation2012; DeVore and Scott Citation2012). Comparisons were made with crystallographic CYP2A6-coumarin complex (PDB-ID: 1Z10) (Yano et al. Citation2005). Before docking, original ligands were deleted and A-chain of each structure was superimposed with VERTAA in BODIL molecular modelling software (Lehtonen et al. Citation2004). Hydrogens were added to each CYP2A13 structure using REDUCE (ver. 3.23) (Word et al. Citation1999). PLANTS software was used for docking with chemPLP scoring function and speed1 setting for search speed (Korb et al. Citation2009). Coordinate for binding site centre was obtained from a central atom of the ligand present in 4EJI, and binding site radius was set to 14 Å. Ten best-scored docking poses for each compound were acquired from each docking, using cluster rmsd 2.5 Å. Scoring functions are known to often fail in identification of the high affinity binding mode. In addition, the highest affinity binding mode is not always equivalent to the catalytically active binding mode. Thus, the following criteria was employed to filter out poses that did not fit to the experimental data of the active molecules: 1. Docking conformations where 7-hydroxylation site was outside radius of 6 Å from the haem iron were discarded using sdfconf (v. 0.8.37; Hritz et al. Citation2008). 2. 7-Hydroxylation site reactivity was considered by discarding conformations where the 7-hydroxylation site was oriented away from the haem group (Sheng et al. Citation2014). 3. Finally, conformations where polar atoms of the coumarin core formed unfavourable contacts with hydrophobic residues instead of forming hydrogen bond were discarded by visual inspection. Formation of at least one hydrogen bond between CYP2A13 and the substrate was required. Docking results were visually inspected in BODIL. Poses where 7-hydroxylation site of the docked compound was oriented away from the haem group were discarded. Due to the relatively small size of the CYP2A13 active site cavity, the remaining docking complexes were subjected to a brief energy minimisation to allow slight relaxation of the active site residues. Minimisation was performed with PRIME module in MAESTRO using VSGB solvation model and OPLS3e force field (Jacobson et al. Citation2002; Citation2004; Li et al. Citation2011; Harder et al. Citation2016). During minimisation, ligand and residues within 5 Å of the ligands were considered flexible. The refinement optimised the docking poses, especially in case of the bulkier 3-phenyl containing compounds. Finally, minimised docking complexes were visually inspected in BODIL to identify active ligand binding modes that were consistent with the hydroxylation reaction taking place at the 7-position of the coumarin core.
Structural figures ( and ) were generated using Pymol 2.3.0 (Schrödinger Citation2015).
Results
Oxidation characteristics
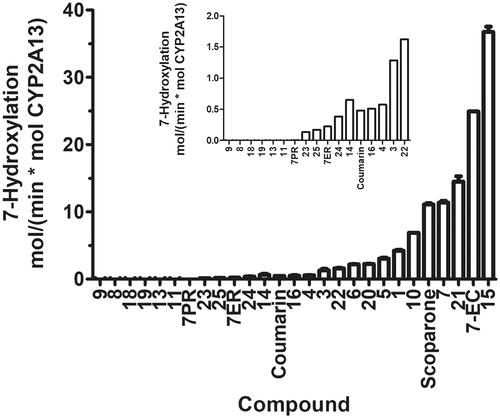
Rates of CYP2A13 catalysed 7-hydroxylation of the coumarins and 7-dealkylation of the resorufins are shown in . Six coumarin derivatives (9, 8, 18, 19, 13, 11) were not oxidised to fluorescent 7-hydroxyl products. The oxidation rate of 14 derivatives (16, 4, 3, 22, 6, 20, 5, 1, 10, scoparone, 7, 21, 7-ethoxycoumarin, 15) was higher than the rate of coumarin 7-hydroxylation, while the rate of four derivatives (23, 25, 24, 14) and both resorufins was lower. The highest rates were observed for 15 (36 min−1), 7-ethoxycoumarin (25 min−1), 21 (14 min−1), 7 (11 min−1) and scoparone (11 min−1). These results indicate that the rate of 7-hydroxylation of coumarins by CYP2A13 is greatly affected by the substituents, and that 7-ethoxyresorufin and 7-pentoxyresorufin are poor substrates for CYP2A13.
Figure 2. Oxidation rates of the coumarin derivatives to fluorescent 7-hydroxy metabolites and 7-dealkylation of 7-ethoxyresorufin and 7-pentoxyresorufin by CYP2A13. Formation of fluorescent metabolites was determined in incubations containing 25 nM CYP enzyme, 20% NADPH regenerating system and 10 µM coumarin derivatives or 1 µM resorufin derivatives in 100 mM Tris-HCl, pH 7.4. The inset represents the 17 lowest oxidation values of the substrates. The activities were determined from the linear phase of the reactions and do not represent Vmax values. 7-EC, 7-ethoxycoumarin.
The enzyme kinetic parameters Km, Vmax and intrinsic clearance (Vmax/Km) were determined for CYP2A13 catalysed oxidation of 16 coumarin derivatives (, Supplement Figure 1). The Km values varied 114-fold (0.9 (4) to 97 (22) µM), Vmax values varied 240-fold (0.25 (4) to 60 (15) mol/(min*mol CYP2A13)) and intrinsic clearance varied 238-fold (26 to 6190 kL/(min*mol CYP2A13)). Oxidation of nine coumarin derivatives was more efficient (higher Vmax/Km ratio) than coumarin 7-hydroxylation, and oxidation of six derivatives was less efficient. The Km values of 3, 4 and 5 were 0.7–1.8 times and 7-ethoxycoumarin, 6, 16 and 21 2.6–5.5 times the Km of coumarin, indicating higher affinity to CYP2A13 than the other compounds. The large variation in the kinetic parameters implies that the substituents markedly affect binding affinity and 7-hydroxylation oxidation efficiency of the coumarin derivatives.
Table 1. The Michaelis-Menten kinetic constants of the 7-hydroxylation of the coumarin derivatives oxidised by human CYP2A13.
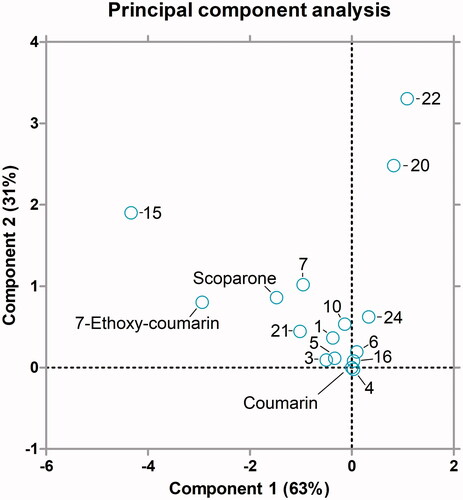
No linear correlation existed between reciprocal values of Km and intrinsic clearance or Vmax. The principal component analysis showed that coumarin and derivatives 1, 3, 4, 5, 6, 10, 16, 21 and 24 formed a basal group of compounds that showed similar kinetic values with coumarin (). Other derivatives branched out to two main directions based on the kinetic parameters. Extremes of these branches were 22 and 20, which had high Km and low Vmax and intrinsic clearance, and 15 and 7-ethoxycoumarin, which had high Vmax and intrinsic clearance. Scoparone and 7 were between the basal group and the extremes with intermediate values of Km, Vmax and Vmax/Km.
Figure 3. Principal component analysis biplot of the Michaelis-Menten kinetic parameters of oxidation of coumarin derivatives by CYP2A13. The analysis was done for the substrates in . The X-axis component explained 63% and the y-axis component 31% of the variation in the data.
Coumarin derivatives poorly oxidised by CYP2A13 such as 22, 20, 6 and coumarin were more efficiently oxidised by CYP2A6 (, Juvonen et al. Citation2019). Compound 4 was equally efficiently oxidised by both CYP2A forms. CYP2A13 oxidised efficiently several coumarin derivatives such as 10, 1, 5, 7, 3, 21, scoparone, 15 and 7-ethoxycoumarin, which are poorly or not at all oxidised by CYP2A6 (, , Juvonen et al. Citation2019).
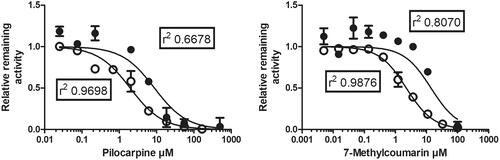
Pilocarpine is an inhibitor of both CYP2A13 and CYP2A6, and 7-methylcoumarin is an inhibitor of CYP2A6 (Kinonen et al. Citation1995; DeVore et al. Citation2012; Juvonen et al. Citation2016). We tested the ability of these inhibitors to suppress CYP2A13 catalysed 7-hydroxylation of coumarin and compound 15 (). The derived IC50 value of pilocarpine was 8.7 µM (95% confidence limit 0–24 µM) for coumarin 7-hydroxylation and 2.0 µM (1.4–2.5 µM) for 7-hydroxylation of 15. IC50 of 7-methylcoumarin was 14 µM (3.5–25 µM) for coumarin 7-hydroxylation and 2.2 µM (1.9–2.6 µM) for 7-hydroxylation of 15. IC50 values of both inhibitors were lower for 15 than for coumarin, as the Km of 15 is higher than that of coumarin.
Figure 4. Inhibition by pilocarpine and 7-methylcoumarin of CYP2A13 catalysed coumarin 7-hydroxylation (closed circles) and 7-hydroxylation of compound 15 (open circles). X-axis indicates concentrations of the inhibitors were added to incubations containing 10 nM enzyme (coumarin) or 2.5 nM enzyme (15), 20% NADPH regenerating system and 10 µM coumarin or 15 in 100 mM Tris-HCl pH 7.4.
Modelling
Molecular docking of compounds 15, 21, 3, 7, 5, 1, 10, 7-ethoxycoumarin, coumarin and scoparone was performed to identify their active binding modes for 7-hydroxylation by CYP2A13. Distance-, orientation- and hydrogen bonding-based filters were used to discard docking poses not consistent with the compound activity data. Poses where 7-hydroxylation site was within 6 Å and orientated towards the haem group and whose binding was stabilised by at least one hydrogen bond were retained: these are referred to as ‘active binding modes’. Docking was performed to all available CYP2A13 crystal structures (PDB: 2P85, 3T3S, 4EJG, 4EJH and 4EJI) to consider flexibility of the active site in identification of active substrate binding modes. Superimposition of the structures showed largest conformational variation for the residues Phe107, Phe118, Phe209, Phe300, Thr305, Met365, Leu366 and Leu370 (Supplement Figure 2(A)). In addition, 4EJI differs from other structures due to conformational change of residues Gln104 and Phe480, which facilitates opening of a possible ligand entry and/or exit channel (Devore and Scott Citation2012; Supplement Figure 2(B)). Docking to 2P85, 3T3S, 4EJG and 4EJH identified at least one active binding mode for 15, 21, 3, 7, 5, 1, 7-ethoxycoumarin, coumarin and scoparone (Supplement Table 1). Generally, poses with the shortest distance between 7-hydroxylation site and haem iron were consistent between docking experiments with different CYP2A13 structures (Supplement Figure 3). Docking with 4EJH showed the best performance, as an active binding mode was ranked first by docking score for five compounds. With 4EJI, docking produced active binding modes for 7-ethoxycoumarin, 21, 3 and scoparone but failed to find suitable poses for other compounds. Many of the compounds were docked into the additional channel of 4EJI active site, which resulted in such poses failing to pass the 6 Å distance filter. Conformation of 4EJI active site may not be relevant in binding prediction of substrates that occupy the area above the haem group but not the additional channel. Docking and minimisation to 3T3S was the only to produce an active binding mode for compound 10 (Supplement Figure 3). Large size of 10 generally caused overlap with the active site residues, which suggests that binding of 10 induces structural adaptation of the CYP2A13 active site that is not visible in the available crystal structures.
All the active compounds were predicted to form hydrogen bond with Asn297 via the carbonyl oxygen of the coumarin core. In each case, binding was stabilised by placement of the coumarin core or a 3-phenyl substituent to the phenylalanine-rich top region of the CYP2A13 active site (). 7-Ethoxycoumarin and scoparone had elevated positioning of the core structure in relation to haem when compared with the other compounds. The terminal CH3 of 7-ethoxycoumarin packed with Leu370 while side chains of Leu366, Thr305 and Ala301, further stabilised positioning of the ethoxy group via hydrophobic contacts (). Hydrophobic packing with the shorter 7-methoxy substituent of scoparone was not as compact and it adopted varying conformations, which could explain the compound’s higher Km (Supplement Figure 3; ). Coumarin, which lacks 7- and 3-position substituents, was positioned closer to haem, resulting in decreased π stacking within the phenylalanine-rich region ().
Figure 5. Molecular docking of 7-ethoxycoumarin and coumarin to CYP2A13. A) Predicted binding mode of 7-ethoxycoumarin with CYP2A13. B) Predicted binding mode of coumarin with CYP2A13. C) Comparison of 7-ethoxycoumarin and coumarin predicted binding modes when complexed with CYP2A13. 7-Ethoxycoumarin and coumarin are shown as sticks. Haem and residues of coumarin-bound, energy minimised CYP2A13 have transparent representation. D) Comparison of coumarin orientation when complexed with CYP2A13 or CYP2A6 ( crystal structure PDB: 1Z10). CYP2A6 residues have transparent representation and residue labels (CYP2A13/CYP2A6) show amino acid differences between 2A13 and 2A6. Ligand, haem and protein atoms and bonds are displayed as sticks. In A and B, dashed line represents a hydrogen bond.
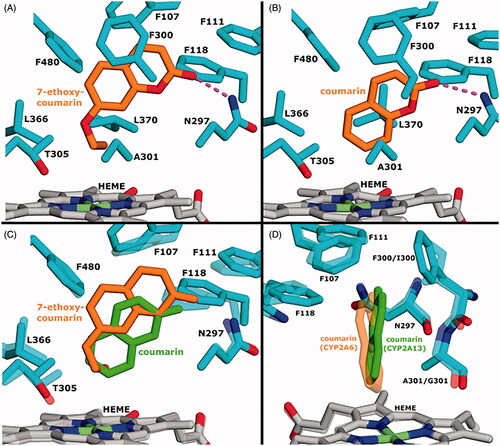
Comparison of the predicted CYP2A13 and crystallographic CYP2A6 coumarin binding modes showed different orientation of the 7-hydroxylation site in relation to haem iron (). In CYP2A13, steric hindrance by the side chain of Ala301 forces coumarin into an orientation where the 7-hydroxylation site points slightly away from the haem iron, explaining the decreased 7-hydroxylation efficiency. Gly301 in CYP2A6 leaves more space into this area and lets coumarin adopt a pose that brings 7-hydroxylation site closer to haem iron. Regarding 7-ethoxycoumarin and scoparone, 7-ethoxy and 7-methoxy substituents are flexible and enable placement of the bulky and rigid core structure to the phenylalanine-rich region, thus fitting better to the proximity of haem next to Ala301 in CYP2A13. This elevated positioning of 7-ethoxycoumarin is not equally well stabilised by π stacking in CYP2A6 due to Phe300 of CYP2A13 being replaced by Ile300. In addition, less compact packing with Gly301 could result in more fluctuation of the 7-ethoxy group, contributing to the decreased reaction efficiency by CYP2A6.
The core structure of the 3-phenyl containing compounds (15, 21, 3, 7, 5, 1, 10) was generally placed closer to haem than that of 7-ethoxycoumarin and scoparone. Compound with the highest Vmax (15) had a binding mode where positioning of the 7-hydroxylation site was stabilised by electrostatic interaction between 6-hydroxyl and haem nitrogens (; ). In addition, the backbone carbonyl oxygen of Leu241 or Leu296 was within reach to form a direct or water-mediated hydrogen bond with 3-hydroxyphenyl in the top region of the binding site (). These hydrogen bond acceptors are less accessible for 4-hydroxyphenyl, which explains the higher Km for compounds carrying hydroxyl or other polar substituents on the 3-phenyl-4-position (15 vs 7, 4 vs 6, 24 and 20; ; ). Proximity of Leu241/296 backbone oxygens is also the likely cause for significant increase in Km when 3- or 4-position of 3-phenyl is occupied by an acetate group (20 and 22). Accordingly, hydrophobic substituents at the 3-phenyl-4-position express lower Km due to hydrophobic contacts with the surrounding residues Leu241, Leu110, Leu296 and Leu244. Predicted binding of 21, 3 and 1 reflect this well as each compound has a halogen-containing substituent at the 3-phenyl-4-position (). However, coupling 3-phenyl-4-trifluoromethoxy with 7-methoxy on the coumarin core abolished 7-hydroxylation activity (18). This could be caused by not having enough space for 4-trifluoromethoxy in the top region due to 7-methoxy elevating the core position. In addition, compounds with methoxy group or fluorine at the 3-phenyl-3-position (9, 8, 19 and 13, respectively) showed very low activity, which could result from bad contacts with Leu241/296 backbone oxygens. 3-Pyridyl of 5 fits well to the phenylalanine-rich region but the compound’s low Vmax, despite carrying 6-hydroxyl, indicates the crucial role of 3-phenyl substituents in stabilisation of an active binding mode. This was indicated also by slight variation in localisation of the 7-hydroxylation site of 5 in the predicted binding modes with different CYP2A13 structures (Supplement Figure 4).
Figure 6. Molecular docking of 3-phenyl containing coumarin derivatives to CYP2A13. A) Predicted binding mode of 15. B) Predicted active binding mode of 21. C) Predicted alternative binding mode of 21. Ligand, protein and haem C atoms are coloured orange, cyan and grey, respectively. Other elements are coloured as follows: O red, N blue, Fe lime, S yellow, F pink and polar H of 15 white. Dashed lines display electrostatic interactions between ligand and protein/haem. In A, two hydrogen bonds between 3-phenyl-3-OH and L241 or L296 are shown that could be formed either directly or through a water molecule.
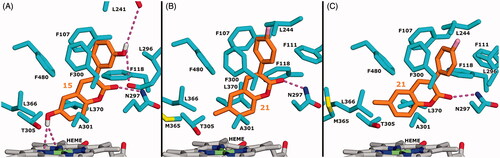
Compounds with 6-methyl, 6-methoxy and 6-chloro substituents (21, 3, 1, 4 and 6) had low Vmax when compared to 15 and 7 that carry 6-hydroxyl. Furthermore, 23, an analogue of 15 with no substituent at the 6-position, showed very low activity, thus indicating the importance of the 6-substituent in anchoring the molecule into the active binding mode. Despite not forming stabilising interaction with haem nitrogens, 6-methyl or 6-methoxy could pack with Met365 to gain support for orientating the 7-hydroxylation site towards haem iron, as shown by predicted binding mode of 21 (). However, CYP2A13 crystal structures show conformational variation for Met365, suggesting that long-lasting stabilisation of the 6-methyl, 6-methoxy and 6-chloro compounds into an active pose could be reduced by the fluctuation of the residue. Met365 conformation in PDB structures 3T3S, 4EJG, 4EJH and 4EJI creates a crevice between Thr305 and Leu366 (DeVore et al. Citation2012; DeVore and Scott Citation2012) () that can be utilised by 6-position substituents. Hydrophobic interactions formed by 6-methyl, 6-methoxy and 6-chloro within the Thr305-Leu366 crevice contribute to binding affinity and explain low Km of such compounds (21, 3, 1, 4, 6) but also cause a less optimal orientation of the 7-hydroxylation site (). In addition, a bulky 6-position substituent, such as 6-methoxy of 1 and 3, can decrease Vmax by sterically hindering accessibility of the 7-hydroxylation site.
Discussion
Lung CYP enzymes oxidise inhaled xenobiotics to more water-soluble and excretable metabolites, but sometimes oxidation leads to reactive, toxic, or carcinogenic metabolites. Procarcinogenic tobacco-derived nitrosamines and polycyclic aromatic hydrocarbons are oxidised to DNA reactive metabolites by CYPs in lung such as CYP2A13 and CYP1s (Hukkanen et al. Citation2002; Anttila et al. Citation2011; Oesch et al. Citation2019). CYP2A13 is mainly expressed in the respiratory tract and plays an important role in the metabolic activation of especially tobacco-derived procarcinogens (Raunio and Rahnasto-Rilla Citation2012; Su and Ding Citation2004; Jalas et al. Citation2005). To gain more insight into the catalytic properties of CYP2A13, we determined CYP2A13 mediated oxidation kinetics of 23 coumarin derivatives and modelled interactions between the ligands and the enzyme active site. CYP2A13 oxidised 18 of 23 nonfluorescent coumarin derivatives to fluorescent 7-hydroxycoumarins with widely varying enzyme kinetic parameters. The active site of CYP2A13 accepted more diversified types of coumarin substrates than the hepatic CYP2A6 enzyme.
Based on Km values, affinity of compounds 3, 4, 5 to the CYP2A13 active site was comparable with the affinity of coumarin. The optimal substituents for binding to the CYP2A13 active site were chloro (4 and 5) or methoxy (3) at position 6, and 3-hydroxyphenyl (4), 4-trifluoromethylphenyl (3) or pyridine (5) at position 3 of the coumarin core. Binding to the CYP2A13 active site was decreased most by triazole (24) or 3-acetylphenyl (22) or 4-acetate phenyl (20) substituents at position 3 of the coumarin. Methoxy at positions 6 (13, 19) or 7 (9, 18) with simultaneous 3-methoxyphenyl (9, 19), 4-trifluoromethoxyphenyl (18) or 3-fluoro-4-hydroxyphenyl (13) at position 3 of coumarin abolished their oxidation. Thus, the type of substituents at positions 6 and 3 of the coumarin core determined the affinity of derivatives to CYP2A13.
Docking of selected derivatives helped to explain the observed variations in substrate binding. Bulkier coumarin derivatives were placed closer to haem than small ones. Carbonyl oxygen of the coumarin core and substituents at positions 3 and 6 were key determinants of binding interactions in the enzyme active site. Carbonyl oxygen formed a hydrogen bond with Asn297, and 3-hydroxyphenyl at position 3 and hydroxyl at position 6 of coumarin anchored the substrates to the CYP2A13 active site. Similarly, a hydrogen bond is formed between carbonyl oxygen of pilocarpine and Asn297 of CYP2A13 (DeVore et al. Citation2012). Compound 15 and 7-ethoxycoumarin were oxidised most efficiently by CYP2A13 and branched out as their own group in the principal component analysis of the kinetic parameters. The ethoxy group of 7-ethoxycoumarin, the compound with the highest intrinsic clearance, was stabilised by hydrophobic packing of the terminal CH3 with surrounding amino acid residues. Packing was less efficient for scoparone having a 7-methoxy group. For compound with the highest Vmax (15), the 7-hydroxylation site was stabilised by electrostatic interactions between 6-hydroxyl and haem nitrogen atoms. For further stabilisation, direct or water-mediated hydrogen bonds with 3-hydroxyphenyl of 15 were formed at the top of the enzyme binding site. These hydrogen bonds were not formed with 4-hydroxyphenyl, which explains the higher Km for compounds with hydroxyl or other polar substituents on the 3-phenyl-4-position (7). Notably, although not suggested by docking, an 180° flip of the 3-hydroxyphenyl would enable hydrogen bonding with Ser208 which has been previously associated with CYP2A13 mediated metabolism of nicotine-derived procarcinogens (He et al. Citation2004). Binding mode of 21 (3-(4-fluorophenyl)-6-methyl-coumarin) suggested that, despite not forming stabilising interaction with haem nitrogen atoms, 6-methyl or 6-methoxy could pack with Met365 to gain support for orientating the 7-hydroxylation site towards haem iron.
Coumarin is the prototype substrate of both CYP2A13 and CYP2A6, and coumarin 7-hydroxylation is a marker activity for these enzymes (Fukami et al. Citation2007; Raunio and Rahnasto-Rilla Citation2012). CYP2A6 was somewhat more efficient in coumarin 7-hydroxylation than CYP2A13, as the Vmax by CYP2A13 was about twofold lower than that of CYP2A6, and the CYP2A13 Km was twofold higher. The observed Vmax and Km for CYP2A13 mediated coumarin 7-hydroxylation are in line with those reported earlier (von Weymarn and Murphy Citation2003; He et al. Citation2004; Su and Ding Citation2004). Docking analyses showed that in CYP2A13 steric hindrance by Ala301 orients coumarin slightly away from haem iron. Gly301 in CYP2A6 lets coumarin adopt a pose that brings 7-hydroxylation site closer to haem iron.
7-Ethoxycoumarin is a substrate for several human CYP enzymes. CYP2A13 catalysed 7-O-deethylation of 7-ethoxycoumarin 1670 times more efficiently than CYP2A6. 7-Hydroxylation of compound 15 by CYP2A13 occurred with the same efficiency as 7-O-deethylation of 7-ethoxycoumarin. The ethoxy substituent in 7-ethoxycoumarin is flexible resulting in placement of the bulky core structure to the phenylalanine-rich region, thus fitting better to the proximity of haem in CYP2A13. This elevated positioning of 7-ethoxycoumarin is not equally well stabilised by π stacking in CYP2A6. In addition, less compact packing with Gly301 in CYP2A6 could result in more fluctuation of the 7-ethoxy group, contributing to the reduced oxidation efficiency of 7-ethoxycoumarin.
Conclusions
We evaluated human CYP2A13 mediated 7-hydroxylation characteristics of 23 coumarin derivatives and dealkylations of 7-ethoxy- and 7-penthoxyresorufins. New sensitive and convenient profluorescent substrates of CYP2A13 were identified, such as high-affinity 6-chloro-3-(3-hydroxyphenyl)-coumarin (4) and high intrinsic clearance 6-hydroxy-3-(3-hydroxyphenyl)-coumarin (15). 6-chloro or 6-methoxy and 3-(3-hydroxyphenyl) or 3-(4-trifluoromethylphenyl) substituents of coumarin increased binding affinity at the active site of CYP2A13, whereas 3-triazole or 3-(3-acetate phenyl) or 3-(4-acetate phenyl) substituents decreased it. The active site of CYP2A13 accepts more diversified types of substrates than CYP2A6. The molecular docking results of this study provided detailed information about interactions between coumarin derivatives and the active site residues of CYP2A13 and explained the enzyme kinetic differences in 7-hydroxylation oxidation among the tested compounds.
Supplemental Material
Download PDF (899.6 KB)Disclosure statement
The authors report no declaration of interest.
References
- Anttila S, Raunio H, Hakkola J. 2011. Cytochrome P450-mediated pulmonary metabolism of carcinogens: regulation and cross-talk in lung carcinogenesis. Am J Respir Cell Mol Biol. 44(5):583–590.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28(1):235–242.
- DeVore NM, Meneely KM, Bart AG, Stephens ES, Battaile KP, Scott EE. 2012. Structural comparison of cytochromes P450 2A6, 2A13, and 2E1 with pilocarpine. FEBS J. 279(9):1621–1631.
- DeVore NM, Scott EE. 2012. Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone binding and access channel in human cytochrome P450 2A6 and 2A13 enzymes. J Biol Chem. 287(32):26576–26585.
- Fernandez-Salguero P, Hoffman SM, Cholerton S, Mohrenweiser H, Raunio H, Rautio A, Pelkonen O, Huang JD, Evans WE, Idle JR. 1995. A genetic polymorphism in coumarin 7-hydroxylation: sequence of the human CYP2A genes and identification of variant CYP2A6 alleles. Am J Hum Genet. 57(3):651–660.
- Fukami T, Nakajima M, Sakai H, Katoh M, Yokoi T. 2007. CYP2A13 metabolizes the substrates of human CYP1A2, phenacetin, and theophylline. Drug Metab Dispos. 35(3):335–339.
- Gonzalez FJ, Coughtrie M, Tukey RH. 2017. Drug metabolism. In: Bruton L, Lazo J, Parker K, editors. Goodman & Gilmans’s the pharmacological basis of therapeutics. 13th ed. New York: McGraw-Hill; p. 85–100.
- Gundert-Remy U, Bernauer U, Blömeke B, Döring B, Fabian E, Goebel C, Hessel S, Jäckh C, Lampen A, Oesch F, et al. 2014. Extrahepatic metabolism at the body's internal-external interfaces. Drug Metab Rev. 46(3):291–324.
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, et al. 2016. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput. 12(1):281–296.
- He XY, Shen J, Ding X, Lu AY, Hong JY. 2004. Identification of critical amino acid residues of human CYP2A13 for the metabolic activation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific carcinogen. Drug Metab Dispos. 32(12):1516–1521.
- He XY, Shen J, Hu WY, Ding X, Lu AY, Hong JY. 2004. Identification of Val117 and Arg372 as critical amino acid residues for the activity difference between human CYP2A6 and CYP2A13 in coumarin 7-hydroxylation. Arch Biochem Biophys. 427(2):143–153.
- He XY, Tang L, Wang SL, Cai QS, Wang JS, Hong JY. 2006. Efficient activation of aflatoxin B1 by cytochrome P450 2A13, an enzyme predominantly expressed in human respiratory tract. Int J Cancer. 118(11):2665–2671.
- Hritz J, de Ruiter A, Oostenbrink C. 2008. Impact of plasticity and flexibility on docking results for cytochrome P450 2D6: a combined approach of molecular dynamics and ligand docking. J Med Chem. 51(23):7469–7477.
- Hukkanen J, Jacob P, Benowitz NL. 2005. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 57(1):79–115.
- Hukkanen J, Pelkonen O, Hakkola J, Raunio H. 2002. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol. 32(5):391–411.
- Jacobson MP, Friesner RA, Xiang Z, Honig B. 2002. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 320(3):597–608.
- Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. 2004. A hierarchical approach to all-atom protein loop prediction. Proteins. 55(2):351–367.
- Jalas JR, Hecht SS, Murphy SE. 2005. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem Res Toxicol. 18(2):95–110.
- Ji M, Zhang Z, Li N, Xia R, Wang C, Yu Y, Yao S, Shen J, Wang SL. 2018. Identification of 5-hydroxymethylfurfural in cigarette smoke extract as a new substrate metabolically activated by human cytochrome P450 2A13. Toxicol Appl Pharmacol. 359:108–117.
- Juvonen RO, Ahinko M, Huuskonen J, Raunio H, Pentikäinen OT. 2019. Development of new Coumarin-based profluorescent substrates for human cytochrome P450 enzymes. Xenobiotica. 49(9):1015–1024.
- Juvonen RO, Ahinko M, Huuskonen J, Raunio H, Pentikäinen OT. Substrate selectivity of coumarin derivatives by human CYP1 enzymes: in vitro enzyme kinetics and in silico modelling. Submitted
- Juvonen RO, Kuusisto M, Fohrgrup C, Pitkänen MH, Nevalainen TJ, Auriola S, Raunio H, Pasanen M, Pentikäinen OT. 2016. Inhibitory effects and oxidation of 6-methylcoumarin, 7-methylcoumarin and 7-formylcoumarin via human CYP2A6 and its mouse and pig orthologous enzymes. Xenobiotica. 46(1):14–24.
- Kinonen T, Pasanen M, Gynther J, Poso A, Järvinen T, Alhava E, Juvonen RO. 1995. Competitive inhibition of coumarin 7-hydroxylation by pilocarpine and its interaction with mouse CYP 2A5 and human CYP 2A6. Br J Pharmacol. 116(6):2625–2630.
- Korb O, Stützle T, Exner TE. 2009. Empirical scoring functions for advanced protein-ligand docking with plants. J Chem Inf Model. 49(1):84–96.
- Lehtonen JV, Still DJ, Rantanen VV, Ekholm J, Björklund D, Iftikhar Z, Huhtala M, Repo S, Jussila A, Jaakkola J, et al. 2004. BODIL: a molecular modeling environment for structure–function analysis and drug design. J Comput Aided Mol Des. 18(6):401–419.
- Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA. 2011. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins. 79(10):2794–2812.
- Li L, Carratt S, Hartog M, Kovalchik N, Jia K, Wang Y, Zhang QY, Edwards P, Winkle LV, Ding X. 2017. Human CYP2A13 and CYP2F1 mediate naphthalene toxicity in the lung and nasal mucosa of CYP2A13/2F1-humanized mice. Environ Health Perspect. 125(6):067004.
- Murphy SE, Raulinaitis V, Brown KM. 2005. Nicotine 5'-oxidation and methyl oxidation by P450 2A enzymes. Drug Metab Dispos. 33(8):1166–1173.
- Niinivehmas S, Postila PA, Rauhamäki S, Manivannan E, Kortet S, Ahinko M, Huuskonen P, Nyberg N, Koskimies P, Lätti S, et al. 2018. Blocking oestradiol synthesis pathways with potent and selective coumarin derivatives. J Enzyme Inhib Med Chem. 33(1):743–754.
- Oesch F, Fabian E, Landsiedel R. 2019. Xenobiotica-metabolizing enzymes in the lung of experimental animals, man and in human lung models. Arch Toxicol. 93(12):3419–3489.
- Parkinson A, Ogilvie BW, Buckley DB, Kazmi F, Parkinson O. 2019. Biotransformation of xenobiotics. In: Casarett LJ, Doull J, Klaassen CD, editors. Casarett & Doull’s toxicology: the basic science of poisons. 9th ed. New York: McGraw Hill; p. 193–430.
- Pelkonen O, Raunio H. 1997. Metabolic activation of toxins: tissue-specific expression and metabolism in target organs. Environmental Health Perspective Suppl. 105(Suppl 4):767–774.
- Rauhamäki S, Postila PA, Niinivehmas S, Kortet S, Schildt E, Pasanen M, Manivannan E, Ahinko M, Koskimies P, Nyberg N, et al. 2018. Structure–activity relationship analysis of 3-phenylcoumarin-based monoamine oxidase B inhibitors. Front Chem. 6:41.
- Raunio H, Rahnasto-Rilla M. 2012. CYP2A6: genetics, structure, regulation, and function. Drug Metabol Drug Interact. 27(2):73–88.
- Schrödinger. 2015. The PyMOL molecular graphics system, Version 2.3.
- Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. 2007. Epik: a software program for pK( a ) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 21(12):681–691.
- Sheng Y, Chen Y, Wang L, Liu G, Li W, Tang Y. 2014. Effects of protein flexibility on the site of metabolism prediction for CYP2A6 substrates. J Mol Graph Model. 54:90–99.
- Shimada T, Takenaka S, Kakimoto K, Murayama N, Lim YR, Kim D, Foroozesh MK, Yamazaki H, Guengerich FP, Komori M. 2016. Structure–function studies of naphthalene, phenanthrene, biphenyl, and their derivatives in interaction with and oxidation by cytochromes P450 2A13 and 2A6. Chem Res Toxicol. 29 (6):1029–1040.
- SIMCA. Umetrics. version 15.0.2. [accessed 2021 Feb 26]. https://www.sartorius.com/en/products/process-analytical-technology/data-analytics-software/mvda-software/simca?gclid=Cj0KCQiAst2BBhDJARIsAGo2ldWk7cPBO2Aqv6npSJGViHKYuCMGMo-H3K2DfuJ-i_hJKkfxdyFSFcgaAnuFEALw_wcB
- Smith BD, Sanders JL, Porubsky PR, Lushington GH, Stout CD, Scott EE. 2007. Structure of the human lung cytochrome P450 2A13. J Biol Chem. 282(23):17306–17313.
- Su T, Ding X. 2004. Regulation of the cytochrome P450 2A genes. Toxicol Appl Pharmacol. 199(3):285–294.
- von Weymarn LB, Murphy SE. 2003. CYP2A13-catalysed coumarin metabolism: comparison with CYP2A5 and CYP2A6. Xenobiotica. 33(1):73–81.
- Word JM, Lovell SC, Richardson JS, Richardson DC. 1999. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 285(4):1735–1747.
- Yano JK, Hsu MH, Griffin KJ, Stout CD, Johnson EF. 2005. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat Struct Mol Biol. 12(9):822–823.
- Zanger UM, Schwab M. 2013. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 138(1):103–141.
- Zhang X, D'Agostino J, Wu H, Zhang QY, von Weymarn L, Murphy SE, Ding X. 2007. CYP2A13: variable expression and role in human lung microsomal metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. J Pharmacol Exp Ther. 323(2):570–578.
- Zhang Z, Yang X, Wang Y, Wang X, Lu H, Zhang X, Xiao X, Li S, Wang X, Wang SL. 2013. Cytochrome P450 2A13 is an efficient enzyme in metabolic activation of aflatoxin G1 in human bronchial epithelial cells. Arch Toxicol. 87(9):1697–1707.