
Abstract
1. NDec is a novel, oral, fixed-dose formulation of decitabine and tetrahydrouridine that is currently being developed for the treatment of patients with sickle cell disease. Here, we examine the potential for both components of NDec to interact with key drug metabolising enzymes (tetrahydrouridine only) and drug transporters (decitabine and tetrahydrouridine).
2. This study assessed the inhibition and induction of cytochrome P450 (CYP) enzymes by tetrahydrouridine, as well as the involvement of specific drug metabolising enzymes in tetrahydrouridine metabolism. Inhibition of efflux and uptake transporters by both decitabine and tetrahydrouridine was also studied.
3. Tetrahydrouridine did not inhibit or induce relevant CYP enzymes at concentrations ranging from 0.1 to 100 μM. Metabolism of tetrahydrouridine did not occur in the presence of the human drug metabolising enzymes tested. Tetrahydrouridine showed weak inhibition towards the MATE2-K transporter (∼30% inhibition at 5 and 50 μM), which was not deemed clinically relevant. Tetrahydrouridine did not inhibit any of the remaining uptake or efflux transporters. Decitabine (0.5 and 5 μM) did not inhibit any of the evaluated uptake or efflux drug transporters.
4. Data presented confirm that tetrahydrouridine and decitabine are unlikely to be involved in metabolism- or transporter-based drug-drug interactions.
Introduction
Sickle cell disease (SCD) is a common inherited blood disorder with a particularly high prevalence in sub-Saharan Africa and India (Piel et al. Citation2017). There are ∼67 000 people affected by SCD in the European Union, and it is estimated that there are around 100 000 people with SCD in the United States (EMA Citation2017; CDC Citation2020). SCD arises due to single nucleotide substitutions in the haemoglobin subunit β gene that cause intracellular polymerisation of deoxygenated sickle haemoglobin (HbS) (Kato et al. Citation2018). This leads to the deformation of erythrocytes into crescent-shaped cells that can cause vaso-occlusive crises (VOCs), haemolytic anaemia and immunoactivation (Manwani and Frenette Citation2013). Resulting health issues in affected patients include acute pain episodes, bacterial infections, acute chest syndrome, stroke, and chronic organ complications (Abboud Citation2020).
Pharmacological reinduction of foetal haemoglobin (HbF) levels after infancy is one treatment approach that can act to substitute HbS in patients with SCD (Steinberg et al. Citation2014; Saunthararajah Citation2019). Hydroxyurea, a ribonucleotide reductase inhibitor that increases HbF expression, was approved for the treatment of SCD by the US Food and Drug Administration (FDA) in 1998, and later by the European Medicines Agency (EMA) in 2007 (Kato et al. Citation2018). Until 2017, when L-glutamine was approved by the FDA, hydroxyurea was the only available treatment for SCD (FDA Citation2017). Voxelotor and crizanlizumab were also subsequently approved by the FDA in 2019, with crizanlizumab being authorised for use in the European Union in 2020 (EMA Citation2021; FDA Citation2019a, Citation2019c). There is currently an unmet medical need for novel treatments that target the root cause of the disease.
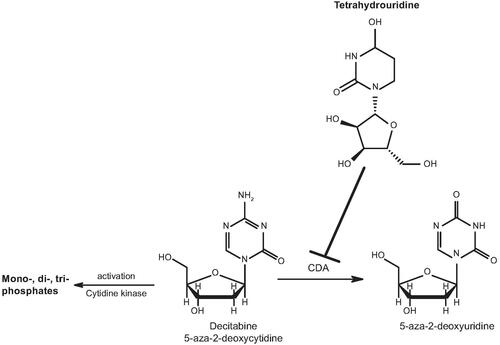
NDec is a novel, oral, fixed-dose combination of decitabine and tetrahydrouridine that is currently under development by Novo Nordisk for the treatment of SCD. NDec is a disease-modifying epigenetic agent that is intended to treat the root cause of SCD through direct elevation of HbF production to prevent haemolytic anaemia and VOCs. In addition, NDec is expected to exhibit an improved tolerability and safety profile when compared with hydroxyurea, allowing patients to remain on treatment for longer. Decitabine, which is the active component of NDec, reinduces HbF expression by inhibiting DNA methyltransferase 1 (DNMT1) activity (Saunthararajah Citation2019). Inhibition of DNMT1 reduces HbF promoter methylation, subsequently increasing HbF gene expression (Charache et al. Citation1983; Paikari and Sheehan Citation2018). Under normal conditions, decitabine is rapidly metabolised by cytidine deaminase (CDA) (), an enzyme with high abundance in the gastrointestinal tract and liver (Lavelle et al. Citation2012; Molokie et al. Citation2017). Tetrahydrouridine is a competitive inhibitor of CDA (Cohen and Wolfenden Citation1971; Beumer et al. Citation2008a; Lavelle et al. Citation2012), and studies in mice and non-human primates confirmed that administration of oral tetrahydrouridine before oral decitabine treatment was able to increase decitabine bioavailability and widen concentration-time profiles (Lavelle et al. Citation2012). While an intravenous formulation of decitabine (Dacogen®) has been approved for the treatment of myelodysplastic syndromes (MDS) in the United States (FDA approval: 2006), and for acute myeloid leukaemia (AML) in Europe (EMA approval: 2012), tetrahydrouridine is not approved for any indication and has no therapeutic effect when used alone. However, tetrahydrouridine has been used previously as a CDA inhibitor in combination with decitabine or cytosine arabinoside in clinical trials (Kreis et al. Citation1991; Marsh et al. Citation1993; Beumer et al. Citation2008b).
Figure 1. Structures of decitabine and tetrahydrouridine. Tetrahydrouridine is an inhibitor of CDA, an enzyme found primarily in the gastrointestinal tract and liver, that metabolises decitabine. CDA: cytidine deaminase.
Due to the severity of SCD, patients may also be prescribed several concomitant medications to treat the various complications of SCD, for example, to alleviate pain (e.g. non-steroidal anti-inflammatory drugs or opiates) and to prevent infection (e.g. antibiotics) (Kato et al. Citation2018). Therefore, it is of high importance to assess the risk of clinical drug-drug interactions (DDIs) for novel SCD treatments. Studies to investigate the DDI potential of new investigational drugs are also required by regulatory bodies during drug development, and relevant guidelines have been developed by the FDA and EMA to guide this process (EMA Citation2012b; FDA Citation2020). The activities of cytochrome P450 (CYP) enzymes, which are essential for hepatic drug metabolism, can be inhibited or enhanced by the presence of perpetrators, potentially resulting in undesired and clinically relevant DDIs. Previous in vitro studies have shown that decitabine does not inhibit CYP1A2, CYP2C8, CYP2C19, CYP2D6, CYP3A4, and CYP2C9, or induce CYP1A2, CYP2B6, CYP2C9, CYP3A4/5, and CYP2E1 at clinically relevant concentrations (EMA Citation2012a). However, neither the CYP inhibition nor induction potential of tetrahydrouridine has so far not been reported. Drug transporters are essential for drug disposition processes within the body and function to limit intestinal absorption and passage across the blood-brain barrier, facilitate excretion into the bile and urine, and regulate accumulation of endogenous and xenobiotic compounds into hepatocytes for further metabolism or excretion into the bile (Giacomini et al. Citation2010; Liang et al. Citation2015). Interference with key drug transporters can therefore increase or decrease drug exposure, resulting in clinically relevant DDIs. No studies have yet been performed to investigate the inhibition of key drug transporters by decitabine or tetrahydrouridine. Therefore, more data are required to further understand the DDI potential of NDec for future clinical development. Additionally, increased knowledge will inform prescribing doctors of the DDI risk and thereby ensure the safety of SCD patients taking co-medications.
Clinical concentrations of decitabine and tetrahydrouridine for the treatment of SCD, which can be used to guide concentration selection in the current in vitro DDI studies, have previously been published as part of a first-in-human trial where a fixed oral tetrahydrouridine dose (10 mg/kg) was administered 60 min before oral decitabine (0.01–0.16 mg/kg) (Molokie et al. Citation2017). The highest measured decitabine concentration in plasma that resulted in DNMT1 depletion and upregulation of HbF was ∼50 nM, which was measured 2 h after the 0.16 mg/kg decitabine dose. However, as pharmacokinetic (PK) sampling was not extensive (i.e. 0, 2, 4, and 24 h after the first decitabine dose), the maximum plasma concentration (Cmax) was not optimally determined from a PK perspective and thus could be underestimated due to the low number of samples. In a recently completed Phase I study designed to investigate the effect of a high-fat, high-calorie meal on the PK of NDec (NCT04086238), healthy adults received similar doses of decitabine and tetrahydrouridine as in the first-in-human study (males: 0.188 mg/kg decitabine and 9.4 mg/kg tetrahydrouridine; females: 0.22 mg/kg decitabine and 11 mg/kg tetrahydrouridine). This resulted in a geometric mean decitabine Cmax of 0.12 µM (male subjects) and 0.13 µM (female subjects). The mean tetrahydrouridine Cmax was ∼5 µM. Unbound Cmax is not expected to differ substantially from total Cmax as plasma protein binding is negligible for decitabine and tetrahydrouridine (Beumer et al. Citation2008a; EMA Citation2019).
The overall objective of this study was to evaluate the risk for clinically relevant DDIs involving drug metabolising enzymes or drug transporters with decitabine and tetrahydrouridine. To support the available data for decitabine, the in vitro inhibitory potential of tetrahydrouridine on the following CYP enzymes was assessed in human liver microsomes (HLMs): CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5. Furthermore, induction of CYP enzyme gene expression and enzyme activities for CYP1A2, CYP2B6, and CYP3A4/5 following exposure of human hepatocytes to tetrahydrouridine was also evaluated. The potential metabolism of tetrahydrouridine by human CYP enzymes, flavin-containing monooxygenase (FMO), uridine diphosphate glucuronosyltransferase (UGT), sulfotransferase (SULT), aldehyde oxidase (AO), and xanthine oxidase (XO) enzymes was investigated in human HLM, S9, and cytosol fractions. Decitabine and tetrahydrouridine were also examined as inhibitors of the following human uptake and efflux drug transporters: Organic Anion Transporter (OAT) 1 and OAT3, Organic Cation Transporter (OCT) 1 and OCT2, Organic Anion Transporting Polypeptide (OATP) 1B1 and OATP1B3, Multidrug and Toxin Extrusion (MATE) 1 and MATE2-K, P-glycoprotein (P-gp), Breast Cancer Resistance Protein (BCRP), and Bile Salt Export Pump (BSEP).
Materials and methods
Experiments were performed by Labcorp Early Development Laboratories Inc (Madison, WI, USA).
Materials
Test articles
Decitabine and tetrahydrouridine were manufactured by ScinoPharm (Tainan, Taiwan).
CYP inhibition studies
Manufacturer details for reagents used in CYP activity assays () are available in Supplementary Methods 1. Pooled HLMs from 150 individuals (79 males and 71 females) were obtained from BioIVT (Baltimore, MD, USA). HLM characterisation information is provided in Supplementary Data 1. Evaluation of microsomal protein binding confirmed that tetrahydrouridine was highly unbound under the experimental conditions described (data not shown).
Table 1. Cytochrome P450 activity assays.
CYP induction studies
Manufacturer details for reagents used in CYP induction studies are available in Supplementary Methods 2. Primary cultures of cryopreserved human hepatocytes from a total of three individual donors (two females and one male) were obtained from BioIVT (Baltimore, MD, USA). Tetrahydrouridine cytotoxicity and stability in hepatocyte cultures were examined and considered acceptable (data not shown). Hepatocyte characterisation information is provided in Supplementary Data 2.
Metabolism of tetrahydrouridine studies
Pooled HLMs from at least 150 donors (both sexes), pooled human liver S9 from at least 10 donors (both sexes), and pooled human liver cytosol from at least 10 donors (both sexes) were obtained from BioIVT (Baltimore, MD, USA). Characterisation information is provided in Supplementary Data 3.
Human drug transporter interaction studies
Manufacturer details for reagents used in drug transporter interaction assays are available in Supplementary Methods 3. Transfected human embryonic kidney (HEK) 293 cell lines were obtained in single-use cryopreserved vials from Corning Life Science (Tewksbury, MA, USA). Caco-2 subclone C2BBe1 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Membrane vesicles from baculovirus-infected insect cells (Sf9) expressing BSEP were obtained from Genomembrane (Thermo Fisher Scientific, Waltham, MA, USA). Solubility, stability, non-specific binding, and cytotoxicity were evaluated and deemed acceptable before conducting drug transporter interaction studies (data not shown).
CYP inhibition studies
Direct inhibition of CYP enzymes by tetrahydrouridine
Details on CYP activity assay substrates, concentrations, incubation times, analytes, and positive controls are provided in . Linear conditions were confirmed before experiments were performed. Eight concentrations of tetrahydrouridine were assessed (0.1, 0.268, 0.720, 1.93, 5.18, 13.9, 37.3, and 100 μM). HLMs (15 μL) were mixed with a substrate (1.25 μL), test article (tetrahydrouridine [2.50 μL] or positive/negative control [1.25 μL]), and assay buffer (0.1 M potassium phosphate buffer containing 1 mM EDTA, pH 7.4; 181.25 μL for tetrahydrouridine or 182.5 μL for positive/negative controls). Mixtures were added to chilled 96-well plates and pre-incubated for 10 min at 37 °C. After pre-incubation, 50 μL pre-warmed nicotinamide adenine dinucleotide phosphate (NADPH, reduced form, 1 mM) was added to initiate the reaction. Reactions were terminated after the time provided in by addition of chilled acetonitrile that contained a stable isotope-labelled internal standard specific to the CYP substrate. Plates were then centrifuged for 5 min at 1000 rpm and supernatants were transferred to a fresh 96-well plate for analysis by liquid chromatography-mass spectrometry (LC-MS). Sample and control incubations were performed in triplicate; standards and quality control samples were performed in duplicate.
Metabolism-dependent inhibition of CYP enzymes by tetrahydrouridine
Preincubation mixtures were first prepared with eight concentrations of tetrahydrouridine (0.1, 0.268, 0.72, 1.93, 5.18, 13.9, 37.3, and 100 μM; predilution concentration), HLMs (10× concentrated) and either NADPH (1 mM) or control solution. After incubation for 30 min at 37 °C, preincubation mixtures (25 μL) were then diluted 10-fold with assay buffer mixture (225 μL) containing NADPH (1 mM) and marker substrate. Incubation conditions were identical to the direct inhibition assay and are described in . Following incubation, assay plates were then centrifuged for 5 min at 1000 rpm. Supernatants were then moved to a separate 96-well plate for LC-MS analysis.
Sample analysis by LC-MS
LC-MS was conducted using a Shimazdu 20 series high-performance liquid chromatograph system (Shimadzu, Kyoto, Japan) and an AB Sciex Triple Quad 5500 mass spectrometry system (Sciex, Framingham, MA, USA). LC-MS parameters are provided in Supplementary Methods 4.
CYP induction studies
Preincubation of hepatocyte culture
Hepatocytes were thawed (90–120 s at 37 °C), transferred to pre-warmed universal cryo recovery medium (UCRM), and centrifuged at 100 × g for 9 min at ambient temperature. The pellet was then resuspended in supplemented Williams’ E plating medium (sWEP) and viability was determined using a trypan blue exclusion assay. The suspension was diluted to a density of 1.4 × 106 cells/mL, and equal volumes of cell suspension and sWEP (50 µL) were added to each well of a 96-well, collagen-coated plate to achieve a final viable cell density of 7.0 × 104 cells/well. The medium was removed after cell attachment and was replaced with 0.1 mL/well of 0.35 mg/mL GelTrex in supplemented Williams’ E maintenance medium (sWEM). Plated hepatocytes were maintained overnight in an incubator at 37 °C (5% CO2, saturated humidity). Morphology and adherence were examined daily.
CYP gene expression analysis
Following overnight acclimation, the culture medium was removed and replaced with tetrahydrouridine (1, 2.5, 5, 10, 25, 50, and 100 µM), prototypical inducer (omeprazole [50 µM], phenobarbital [1000 µM] or rifampicin [10 µM]), non-inducer (methotrexate [20 µM]), or solvent control dosing solution (0.1% DMSO in sWEM; 0.1 mL/well). Dosing was repeated every 24 ± 2 h so that the hepatocytes were exposed for a total of 72 ± 2 h. Tetrahydrouridine and control solutions were then removed, and hepatocytes were washed with Dulbecco’s phosphate-buffered saline, lysed, and mRNA collected using the mRNA Catcher Plus kit (Invitrogen, Carlsbad, CA, USA). The mRNA was transcribed to cDNA on the same day of extraction using TaqMan Reverse Transcription reagents (Applied Biosystems, Foster City, CA, USA). mRNA was stored at −70 °C, and cDNA was stored at −20 °C until further analysis. Relative concentrations of CYP1A2, CYP2B6, and CYP3A4 mRNA were determined by Real-Time Polymerase Chain Reaction using a QuantStudio 6 Flex system (Applied Biosystems), TaqMan Fast Advanced Master Mix (Applied Biosystems), and enzyme-specific probes (Applied Biosystems). For each sample, CYP mRNA was normalised to the mRNA of an endogenous control (glyceraldehyde 3-phosphate dehydrogenase [GAPDH]).
CYP enzyme activity analysis
Tetrahydrouridine (1, 10, and 100 µM), prototypical inducers, and non-inducer was removed after 24 ± 2 h and replaced with sWEM (0.1 mL) containing appropriate probe substrates (CYP1A2: Phenacetin [100 µM]; CYP2B6: Bupropion [500 µM]; CYP3A4/5 Midazolam [30 µM]). Plates were incubated for 2 h, after which reactions were quenched with 0.1 mL acetonitrile. Samples were harvested into new 96-well plates, centrifuged (2000 × g for 5 min), and supernatants were transferred to new 96-well plates. Supernatants were transferred to 96-well analysis plates before analysis, and the appropriate internal standard was added. Supernatant and analysis plates were stored at −20 °C.
Sample analysis by LC-MS
CYP activities were determined by quantifying the production of enzyme-specific metabolites using a Shimazdu 20 series high-performance liquid chromatograph system (Shimadzu, Kyoto, Japan) and an AB Sciex Triple Quad 5000 mass spectrometry system (Sciex, Framingham, MA, USA) (Supplementary Methods 4). Quantification was achieved by comparison to standard curves of the metabolites.
Involvement of drug metabolising enzymes in the metabolism of tetrahydrouridine
HLMs (CYP and FMO enzymes)
HLMs (0.5 mg/mL) and tetrahydrouridine (1 μM) in phosphate assay buffer (100 mM potassium phosphate buffer, pH 7.4, containing 1 mM EDTA) were pre-incubated at 37 °C for 5 min. Reactions were initiated by adding NADPH (reduced form, 1 mM), and following incubation (0 and 60 min), reactions were terminated with ice-cold acetonitrile or other appropriate stopping solution. Separate control incubations were performed in the absence of either HLMs or NADPH. Additional samples were pre-incubated for 30 min with a non-specific CYP inhibitor (1‐aminobenzotriazole [1 mM] and NADPH) before the addition of tetrahydrouridine. Heat-treated HLMs (45 °C for 5 min) were included as a control for FMO enzyme inactivation.
HLMs (UGT enzymes)
HLMs were activated by pre-incubation on wet ice for at least 15 min with alamethicin (50 μg/mg protein). HLMs (0.5 mg/mL) and tetrahydrouridine (1 μM) in Tris assay buffer (50 mM Tris-HCl buffer, pH 7.4, containing 150 mM KCl and 10 mM MgCl2) were pre-incubated at 37 °C for 5 min. The reaction was initiated by the addition of uridine 5′-diphosphate glucuronic acid (UDPGA, 2 mM), and following incubation (0 and 60 min), the reactions were terminated with ice-cold acetonitrile or other appropriate stopping solution. Separate control incubations were performed in the absence of HLMs and UDPGA.
S9 (SULT enzymes)
S9 (2.5 mg/mL) and tetrahydrouridine (1 μM) in phosphate assay buffer were pre-incubated at 37 °C for 5 min before initiation of the reaction with 3′-phosphoadenosine-5′-phosphosulfate (PAPS, 1 mM). Following incubation (0 or 60 min), the reaction was terminated with ice-cold acetonitrile or other appropriate stopping solution. Separate control incubations were performed in the absence of S9 and PAPS.
Cytosol (AO and XO enzymes)
Cytosol (1 mg/mL) in phosphate assay buffer was pre-incubated at 37 °C for 5 min. The reaction was initiated by the addition of tetrahydrouridine (1 μM). Following incubation (0 or 60 min), the reaction was terminated by ice-cold acetonitrile or other appropriate stopping solution. Additional samples were prepared and incubated in the presence of the AO inhibitor menadione (100 μM) and the XO inhibitor allopurinol (100 μM).
Sample analysis by LC-MS
A Shimazdu 20 series high-performance liquid chromatograph system (Shimadzu, Kyoto, Japan) and an AB Sciex Triple Quad 5000 mass spectrometry system (Sciex, Framingham, MA, USA) were used for analysis (Supplementary Methods 4).
Human drug transporter interaction studies
Initial transporter inhibition experiments were performed using two concentrations per test compound: 0.5 and 5 μM decitabine or 5 and 50 μM tetrahydrouridine where the highest test concentration was ≥10-fold the estimated plasma Cmax. A full concentration range enabling IC50 determinations was to be assessed if clinically relevant inhibition effects were detected in the initial screening step.
Inhibition of uptake transporters by decitabine and tetrahydrouridine
HEK293 cells transfected with the appropriate transporter (OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE 1, and MATE2-K) were thawed at 37 °C and resuspended in supplemented Dulbecco’s Modified Eagle’s Medium (sDMEM) containing 10% (v/v) foetal bovine serum and 1% (v/v) non-essential amino acids. Cells were seeded into 24-well poly-D-lysine-treated plates at an appropriate density and incubated at 37 °C for 3–4 h with 5% CO2 and saturated humidity. The media was then replaced with fresh sDMEM, and the cells were kept at 37 °C (5% CO2 and saturated humidity) for ∼24 h. sDMEM contained either 5 mM sodium butyrate (OATP1B1, OATP1B3, and associated vector control cells) or 2 mM sodium butyrate (MATE1, MATE2-K, and associated vector controls). After the culture medium was removed by aspiration, each well was washed once with transport buffer (HBSS with 10 mM HEPES, pH 7.4) and pre-incubated with transport buffer for 15 min (MATE1 and MATE2-K only) or 30 min. Buffer was then removed by aspiration. For MATE1 and MATE2-K, cells were pre-treated with 40 mM ammonium chloride in transport buffer for 20 min at 37 °C, which was followed by aspiration of the buffer. Pre-warmed (37 °C) working solution containing probe substrate (300 μL) was added to each well to initiate uptake and incubated at 37 °C for 2 min in the case of MATE1 and MATE2-K, or 5 min for remaining transporters. The uptake of probe substrates was examined for each transporter in the presence of a vehicle, selective inhibitor, and either 0.5 and 5 μM decitabine or 5 and 50 μM tetrahydrouridine. Uptake was terminated by quickly aspirating the working solution and rinsing the cells three times with 400 μL of ice-cold transport buffer solution. Cells were lysed with 1 N sodium hydroxide (NaOH) at an ambient temperature for at least 10 min with gentle shaking. Cell lysates were then analysed by liquid scintillation counting (LSC) to quantify the radioactivity of probe substrates. Cell cultures were prepared in parallel to measure the amount of protein per well on each day experiments were carried out.
Inhibition of efflux transporters by decitabine and tetrahydrouridine
Caco-2 subclone C2BBe1 cells were maintained at 37 °C (5% CO2 with saturated humidity) in sDMEM containing 10% (v/v) foetal bovine serum, 2 mM glutamine, 1% (w/v) non-essential amino acids, 100 units/mL of penicillin, and 100 μg/mL of streptomycin. Cells were plated in 24-well Co-star Transwell® polyester membrane inserts (pore size 0.4 μm) in sDMEM at an initial density of 2 × 105 cells/mL. To ensure the complete formation of monolayers and full expression of membrane proteins, monolayers were maintained at 37 °C (5% CO2 with saturated humidity) for 24 days. The medium was replaced at least three times per week. Apical to basolateral and basolateral to apical permeability was determined by measurement of trans-epithelial electrical resistance (TEER) with a volt-ohm metre (World Precision Instruments Inc., Sarasota, FL, USA) to confirm the formation of tight junctions. After removal of culture medium by aspiration, transwell inserts were pre-incubated with Caco-2 transport buffer containing vehicle for 30 min (upper and lower compartments). For each transporter, the apparent permeability of the probe substrate was assessed by adding fresh transport buffer containing either vehicle, selective inhibitor, 0.5 and 5 μM decitabine, or 5 and 50 μM tetrahydrouridine to the receiver compartments. To initiate transport, a working solution with or without inhibitor or decitabine/tetrahydrouridine was added to corresponding donor compartments. After samples were incubated at 37 °C for 2 h, donor and receiver samples were collected and analysed by LSC to quantify the radioactivity of probe substrates. To confirm that the monolayers were suitable, the apparent permeabilities of paracellular (mannitol) and transcellular (caffeine) marker compounds and probe substrates were determined.
Inhibition of vesicular uptake by decitabine and tetrahydrouridine
Sf9 membrane vesicles transfected with BSEP were thawed and combined with assay uptake buffer (50 μg protein/well), substrate, inhibitor, and decitabine or tetrahydrouridine. Mixtures were added to a 96-well plate that was preincubated at 37 °C for 5 min. Prewarmed (37 °C) adenosine triphosphate (ATP) solution was added to the appropriate wells to initiate the reaction, and prewarmed adenosine monophosphate (AMP) solution was added to negative control wells (both ATP and AMP were added at a final concentration of 5 μM). The plate was incubated at 37 °C for 30 min. Chilled wash buffer was added to terminate the reaction and the plate was kept on ice. Chilled blocking buffer was added to a filter plate and a vacuum was applied. The filter plate was then washed with chilled wash buffer and the incubation media was transferred to the pre-treated filter plate and a gentle vacuum was applied. The filter plate was washed four times with chilled wash buffer, ethanol was added to the filter plate, and the samples were eluted into a deep-well 96-well plate at ambient temperature for 12 min. Sample radioactivity was determined using LSC.
Sample analysis by LSC
Radiolabelled samples (100 µl) were added to 5 mL of scintillation fluid (Perkin Elmer, Waltham, MA, USA), vortexed to mix, and counted by LSC using a Tri-Carb 2900TR Liquid scintillation analyser (Perkin Elmer, Waltham, MA, USA).
Data acceptance criteria
CYP inhibition studies
Data were considered acceptable in the direct inhibition assay when there was at least 50% inhibition by positive controls. In the metabolism-dependent assay, data were considered acceptable when inhibition by the positive control resulted in normalised + NADPH positive control percentage inhibition values >40%. This was calculated using the following equation: (% Activity Remainingcontrol) − (% Activity Remaining+NADPH) ≥ 40%.
CYP induction studies
Data were considered acceptable when there was ≥4-fold induction of gene expression and ≥2-fold induction of enzyme activity with positive control inducers when normalising to the vehicle control.
Human drug transporter interaction studies
Data in the HEK293 uptake system were considered acceptable when a ≥4-fold uptake of probe substrates was demonstrated over the vector control and when there was >50% inhibition of substrate uptake in the presence of selective inhibitors. Data collected on inhibition of efflux transporters using the Caco-2 efflux system were deemed acceptable when efflux ratios of probe substrates were ≥4 and there was >50% inhibition of probe substrate efflux ratio by selective inhibitors. TEER values were >200 Ω cm2 and the apparent permeability of 14C-mannitol was ≤2 × 10−6 cm/s and 14C-caffeine was ≥20 × 10−6 cm/s. The membrane vesicle assay system was deemed acceptable when there was a ≥4-fold ATP-dependent uptake of probe substrates when compared with AMP controls and >50% inhibition of the uptake of probe substrates by selective inhibitors.
Inhibition criteria
CYP inhibition is defined as <80% remaining activity (relative to the solvent control). Inhibition of uptake transporters and efflux transporters (membrane vesicles) is defined as >25% inhibition of probe substrate uptake. Efflux transporters (Caco-2 cells) are per definition inhibited when there is >25% decrease in efflux ratio of probe substrate.
Induction criteria
For CYP induction studies, normalised mRNA expression or enzyme activity that is increased ≥2-fold and exhibits concentration dependence after tetrahydrouridine exposure is deemed a positive result.
Statistical analysis
Calculations are provided in Supplementary Methods 5. Descriptive statistics, including mean, standard deviation, relative standard deviation, and linear regression analysis, were used where appropriate. Activities were calculated using SoftMax Pro or Microsoft Excel Version 16.0.
Results
Direct inhibition of CYP enzymes
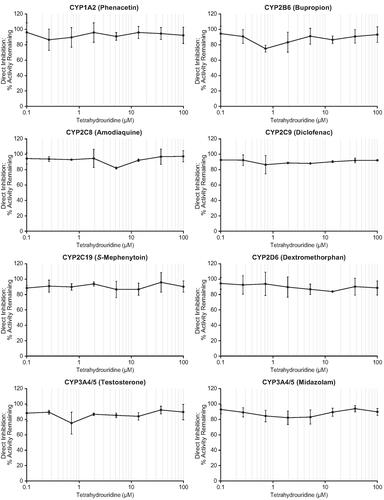
Incubation of HLMs in the presence of tetrahydrouridine at concentrations ranging from 0.1–100 μM did not directly inhibit any of the following CYP enzymes: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5 ( and Supplementary Table 1). Remaining CYP activities at all tetrahydrouridine concentrations (i.e. the CYP enzyme activity remaining after normalisation to solvent control values) were ≥86.3% for CYP1A2, ≥74.6% for CYP2B6, ≥81.6% for CYP2C8, ≥86.3% for CYP2C9, ≥86.6% for CYP2C19, ≥83.6% for CYP2D6, ≥75.0% for CYP3A4/5 (testosterone), and ≥81.9% for CYP3A4/5 (midazolam). While CYP2B6 and CYP3A4/5 both had single activity remaining values below the 80% threshold, which were both measured with 0.72 μM tetrahydrouridine, concentration-dependent inhibition was not observed. The activities of CYP enzymes were inhibited as expected by all positive controls, with activity remaining values ≤24.9%.
Figure 2. Direct inhibition of CYP enzymes by tetrahydrouridine. Shown are mean activity remaining (%) values at eight concentrations of tetrahydrouridine (0.1, 0.268, 0.720, 1.93, 5.18, 13.9, 37.3, and 100 μM). Error bars show ± SD. CYP: cytochrome P450; SD: standard deviation.
Metabolism-dependent inhibition of CYP enzymes
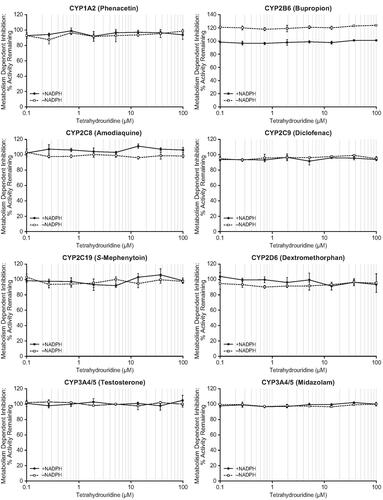
Incubation with tetrahydrouridine did not result in metabolism-dependent inhibition of the tested CYP enzymes ( and Supplementary Table 2). Remaining activity was similar in both + NADPH and − NADPH groups for each of the CYP enzymes tested. Positive controls showed differences in remaining activity ≥47.7% when comparing samples preincubated in the absence and presence of NADPH, which fulfilled the data acceptance criteria.
Figure 3. Metabolism-dependent inhibition of CYP enzymes by tetrahydrouridine. Shown are mean activity remaining (%) values at eight concentrations of tetrahydrouridine (0.1, 0.268, 0.720, 1.93, 5.18, 13.9, 37.3, and 100 μM). Error bars show ± SD. CYP: cytochrome P450; SD: standard deviation.
Induction of CYP enzymes
For analysis of CYP gene expression, human hepatocytes from three donors were exposed to 1, 2.5, 5, 10, 25, 50, and 100 µM tetrahydrouridine for 72 ± 2 h. The maximum fold induction in CYP1A2, CYP2B6, and CYP3A4/5 mRNA levels relative to the solvent control, the tetrahydrouridine concentration at which the maximum value was observed, and the percentage of the positive control are reported in . For each CYP enzyme, increases in mRNA expression were ≥2-fold at some of the tetrahydrouridine concentrations tested. However, increases were not concentration dependent and were comparable to control values obtained with the non-inducer (methotrexate). Overall, these data indicate that tetrahydrouridine is not an inducer of the three CYP enzymes examined.
Table 2. Induction of CYP mRNA and enzyme activities by tetrahydrouridine.
For analysis of CYP enzyme activities, human hepatocytes from three donors were exposed to 1, 10, and 100 µM tetrahydrouridine for 72 ± 2 h. Change in activity was again ≥2-fold for some of the tetrahydrouridine concentrations tested, but changes were not concentration-dependent and were comparable with the effects of the non-inducer (methotrexate).
Involvement of drug metabolising enzymes in the metabolism of tetrahydrouridine
There was no loss (≥85% remaining) of tetrahydrouridine with the HLM, S9, or cytosol fractions under the conditions tested (). CYP, FMO, UGT, SULT, AO, and XO enzymes were therefore not considered to contribute to the metabolism of tetrahydrouridine.
Table 3. Involvement of key drug metabolising enzymes in the metabolism of tetrahydrouridine.
Inhibition of transporters
Full IC50 determinations were not performed in this study as the initial screening experiment showed no relevant inhibition at concentrations ≥10-fold the reported clinical exposure for any of the tested transporters
Inhibition of uptake transporters
Probe substrate uptake by HEK293 cells transfected with OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, or MATE2-K was examined in the presence of two concentrations of decitabine (0.5 and 5 μM). For each of the uptake transporters tested, the mean percent activity remaining values were ≥78.3% (). At tetrahydrouridine concentrations of 5 and 50 μM, probe substrate uptake by MATE2-K was slightly inhibited (activity remaining was 71.7% with 5 μM tetrahydrouridine and 66.5% with 50 μM tetrahydrouridine). The IC50 value for MATE2-K was estimated to be >50 μM. Tetrahydrouridine did not inhibit any of the other transporters (OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, or MATE1), with the remaining activity being ≥81.8% for each. All positive control inhibitors inhibited transporter activity and met the predetermined acceptance criteria.
Table 4. Uptake of probe substrates (HEK293 cells).
Inhibition of efflux transporters (P-gp and BCRP)
The efflux ratio for P-gp-mediated transport of 3H-digoxin was 109 and 127% compared to the solvent control for 0.5 and 5 μM decitabine, respectively (). The efflux ratio for BCRP-mediated transport of 3H-estrone-3-sulfate was 105 and 116% compared to the solvent control for 0.5 and 5 μM decitabine, respectively. Decitabine, therefore, did not inhibit P-gp and BCRP efflux transporters.
Table 5. Permeability and efflux ratios of probe substrates (Caco-2 cells).
The efflux ratio of P-gp-mediated transport of 3H-digoxin was 97.8 and 118% compared to the solvent control for 5 and 50 μM tetrahydrouridine, respectively (). The efflux ratio of BCRP-mediated transport of 3H-estrone-3-sulfate was 102 and 110% compared to the solvent control for 5 and 50 μM tetrahydrouridine, respectively. Tetrahydrouridine was not an inhibitor of P-gp and BCRP efflux transporters.
Inhibition of efflux transporters (BSEP)
Compared to the control, the mean percent remaining activity for 0.5 and 5 μM decitabine was 104 and 96.9%, respectively (). Compared to the control, the mean percent remaining activity 5 and 50 μM tetrahydrouridine was 109 and 103%, respectively. These results show that decitabine and tetrahydrouridine do not inhibit BSEP at the concentrations examined.
Table 6. ATP-dependent uptake of probe substrates (BSEP membrane vesicles).
Discussion
The objective of the current study was to: (1) assess the in vitro CYP inhibition and induction potential of tetrahydrouridine, (2) investigate the potential contribution of drug metabolising enzymes to the metabolism of tetrahydrouridine, and (3) evaluate the in vitro transporter inhibition potential of decitabine and tetrahydrouridine. These in vitro data were used to assess the risk for decitabine and tetrahydrouridine to be involved in clinically relevant metabolism- and transporter-based DDIs. Results showed weak inhibition of tetrahydrouridine against MATE2-K (∼30% inhibition observed at 5 and 50 µM), which was of low clinical relevance considering the observed human exposure. The components of NDec (tetrahydrouridine and decitabine) did not inhibit the remaining drug transporters tested. Tetrahydrouridine did not inhibit or induce the investigated CYP enzymes, and none of the evaluated drug metabolising enzymes was found to be major contributors to the metabolism of tetrahydrouridine.
Decitabine and tetrahydrouridine perpetrator drug properties against CYP enzymes
Dacogen® is an intravenous formulation of decitabine that has been approved to treat MDS and AML in the United States and Europe, respectively. Data on the CYP inhibition and induction potential of decitabine are contained within the Summary of Product Characteristics (SmPC) as well as an EMA public assessment report (EMA Citation2012a; FDA Citation2018). The inhibitory potential of the following CYP enzymes by decitabine was assessed using pooled human liver microsomes: CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. In addition, the induction potential of the following CYP enzymes was examined using cultures of primary human hepatocytes: CYP1A2, CYP2B6, CYP2C9, CYP3A4/5, and CYP2E1. Based on these data, it was concluded that decitabine does not inhibit or induce CYP enzymes at concentrations >20-fold higher than the therapeutic maximum plasma concentration (EMA Citation2012a, Citation2019). These results are informative for NDec as the observed Cmax when decitabine is administered intravenously for MDS and AML is ∼4-fold higher than the observed clinical concentration for oral decitabine in patients with SCD. It should be noted that the decitabine CYP perpetrator profile was examined based on the guidance for DDI studies available at the time of approval. Therefore, evaluation of mechanism‐based inhibition potential, which is recommended in current guidelines, is not described in the product label. Furthermore, the CYP induction study used CYP activity as the endpoint without any assessments related to change in mRNA. The perpetrator risk profile of decitabine against CYP enzymes is however still judged to be low considering not only the previously reported in vitro studies but also the fact that physicians have to date gained ∼15 years of clinical experience with decitabine in the treatment of MDS and AML. This patient group is expected to be taking several concomitant medications, meaning that clinically relevant DDIs would most likely have been detected. As an example, newly diagnosed AML patients can be prescribed decitabine in combination with the CYP3A4 substrate venetoclax (VENCLEXTA®) (FDA Citation2019b), which was shown to be effective and well-tolerated in elderly patients with AML (DiNardo et al. Citation2019). According to the venetoclax FDA product label, concomitant use with strong/moderate CYP3A4 inducers should be completely avoided. Additionally, use of strong/moderate CYP3A inhibitors or P-gp inhibitors requires dose adjustment of venetoclax. The established combination of decitabine and venetoclax highlights the lack of relevant interactions with decitabine and the most clinically relevant CYP enzyme (CYP3A4).
In contrast to the previous perpetrator CYP assessments for decitabine, there were no such data available for tetrahydrouridine. The current study, therefore, investigated the in vitro inhibitory potential (direct inhibition and metabolism-dependent inhibition) of tetrahydrouridine, which showed no inhibition of the tested CYP enzymes in HLMs. Investigations related to tetrahydrouridine CYP induction potential using human hepatocytes were also performed. It was deemed feasible and relevant to use human hepatocyte cultures for these assessments as key transporters (human equilibrative nucleoside transporters [hENTs] and human concentrative nucleoside transporters [hCNTs]) involved in the cellular uptake of endogenous nucleosides and nucleoside analogues are reported to be expressed in the human liver (Govindarajan et al. Citation2007). The assessment of CYP induction potential showed that tetrahydrouridine did not elevate CYP1A2, CYP2B6, and CYP3A4/5 mRNA expression or activity in human hepatocytes beyond background levels. While increases in mRNA expression were ≥2-fold at some of the tetrahydrouridine concentrations evaluated, the same level of induction was also observed with non-inducer control samples. This is likely due to the high induction sensitivity of this hepatocyte lot, and the higher observed control values should be considered as background. Changes were also not concentration dependent, and in general, <20% of the effects observed with positive controls.
Transporter-based drug-drug interactions for decitabine and tetrahydrouridine
Newer regulatory guidelines are now in place since the initial approval of decitabine in 2006, and additional requirements have been included for the assessment of drug transporters (EMA Citation2012b; FDA Citation2020). Analysis of in vitro inhibition of the following drug transporters is recommended by FDA guidelines: P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, MATE1, MATE2-K, and OCT2 (FDA Citation2020). EMA guidelines state that P-gp, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, BCRP should be investigated, and assessment of OCT1, MATE1, and MATE2 should also be considered; inhibition of the transporter BSEP is also recommended by the EMA (EMA Citation2012b). The current study therefore assessed the impact of decitabine and tetrahydrouridine on OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, P-gp, BCRP, and BSEP. Minor limitations with the selected in vitro transporter inhibition study design are noted. For example, it is recommended in the latest FDA guidelines to assess the potential of time-dependent OATP inhibition, which was not investigated in the current study. Furthermore, IC50 values were not estimated as no clinically relevant inhibition was observed at concentrations ≥10-fold the observed plasma Cmax of decitabine and tetrahydrouridine. These limitations will be considered during the further development of NDec, but the risk of clinically relevant DDIs involving drug transporter substrates is considered evidently low based on the observed results.
Nucleoside analogues are polar hydrophilic molecules and therefore require uptake transporters to cross cell membranes; decitabine cellular uptake is reported to be mediated by hENT1 and hENT2 (Damaraju et al. Citation2012; EMA Citation2019). Furthermore, in vitro studies showed that decitabine inhibits hCNT1 and hCNT3 (Damaraju et al. Citation2012; Arimany-Nardi et al. Citation2014). To complement the existing transporter data, the authors are planning to perform in vitro studies investigating the inhibition and substrate potential of tetrahydrouridine towards relevant nucleoside transporters.
Victim drug properties of decitabine and tetrahydrouridine
Decitabine is primarily eliminated via metabolism by CDA, which is found in the liver, kidney, intestinal epithelium, and blood (EMA Citation2012a; Lavelle et al. Citation2012; Molokie et al. Citation2017). The formed metabolites are excreted renally as ∼90% of the administered radiolabelled dose can be found in urine (EMA Citation2012a). In vitro studies showed that CYP enzymes play a minor role in the metabolism of decitabine (EMA Citation2019) and decitabine can therefore be administered safely together with CYP inhibitors and inducers.
In contrast to decitabine, formal in vitro reaction phenotyping studies investigating the potential contribution of drug metabolising enzymes to metabolism of tetrahydrouridine were previously lacking. Data presented in the current study conclude that no major metabolism occurs in the presence of CYP, FMO, UGT, SULT, AO, and XO enzymes. These results are in line with the expectations for nucleoside analogues as they can be envisaged to be substrates for the same metabolic enzymes as endogenous nucleosides, such as kinase, phosphorylase, and deaminase enzymes (e.g. CDA) (Jordheim et al. Citation2013; Varga et al. Citation2016). These results further support that NDec can be administered together with inhibitors and inducers of the major drug metabolising enzymes without any risks of clinically relevant DDIs.
Drug-drug interaction risk for approved SCD drugs
The results of the current study together with the previously published data suggest that the risk of clinically relevant metabolism- and transporter-based DDIs is low for NDec, which is in line with the majority of other approved treatments for SCD. No formal DDI studies have been performed for L-glutamine (Endari™) according to the product label (FDA Citation2017), and CYP and transporter inhibition data are also not described in the label for crizanlizumab-tmca (ADAKVEO®) (FDA Citation2019a). The SmPC for Siklos® mentions that hydroxycarbamide is not metabolised in vitro by CYP enzymes and that it is not a P-gp substrate in vitro (EMA Citation2008). Voxelotor (Oxbryta™) is an HbS polymerisation inhibitor that was approved by the FDA in 2019 for the treatment of SCD (FDA Citation2019c). As described in the FDA label, oxidation of voxelotor is mediated primarily by CYP3A4, and it is consequently recommended to avoid concomitant use with strong CYP3A4 inhibitors and strong or moderate CYP3A4 inducers. Furthermore, treatment of healthy subjects with voxelotor resulted in increased plasma exposure of the CYP3A4 substrate midazolam (1.6-fold) and it is therefore recommended to avoid simultaneous administration with sensitive CYP3A4 substrates. In cases where this is unavoidable, prescribers are recommended to consider reducing the dose of the sensitive CYP3A4 substrate. In vitro DDI data for voxelotor are also briefly mentioned in the label and describe voxelotor as both a reversible and time-dependent inhibitor and inducer of CYP2B6. It is also stated that voxelotor does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, MATE1, MATE2-K, or BSEP. Voxelotor is also not a substrate of P-gp, BCRP, OATP1A2, OATP1B1, OATP1B3, or BSEP.
Conclusion
In summary, decitabine did not inhibit any of the tested transporters, including OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, P-gp, BCRP, and BSEP. Tetrahydrouridine did not show direct or metabolism-dependent inhibition of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5 at concentrations up to 100 μM. Tetrahydrouridine also did not induce expression of CYP1A2, CYP2B6, and CYP3A4/5 in human hepatocytes. Major drug metabolising enzymes were not involved in the metabolism of tetrahydrouridine. While tetrahydrouridine was found to be a weak inhibitor of MATE2-K, the level of inhibition was not sufficient to determine the IC50. The IC50 value was therefore estimated to be >50 μM and subsequently not clinically relevant. Tetrahydrouridine did not inhibit any of the other transporters examined in the present study. Based on the presented data, SCD patients could safely use concomitant medications involving the most common drug metabolising enzymes and drug transporters as there is a low risk of metabolism- and transporter-based DDIs with both components of NDec.
Supplemental Material
Download PDF (376.9 KB)Acknowledgements
The authors would like to acknowledge the study directors (Tyler Wittkopp, Yu He, and Kathleen Sampson) and study coordinators (Mikaela Mullady and Jody Wanta) from Labcorp Early Development Laboratories Inc. (Madison, WI, USA). The authors would also like to thank Signe Beck Petersen, Soraya Benchikh El Fegoun, Inga Bjørnsdottir, Carsten Dan Ley, and Helene Jacobsen, and Ute Friedrich for their critical review of the manuscript. Charlotte Gabel-Jensen is acknowledged for useful scientific discussions.
Disclosure statement
Carolina Säll is an employee of Novo Nordisk A/S. Christian Fogt Hjorth is an employee of Novo Nordisk A/S and holder of Novo Nordisk stocks.
Data availability statement
Data sets for the research presented in the publication are available from the corresponding author upon reasonable request.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Abboud MR. 2020. Standard management of sickle cell disease complications. Hematol Oncol Stem Cell Ther. 13(2):85–90.
- Arimany-Nardi C, Errasti-Murugarren E, Minuesa G, Martinez-Picado J, Gorboulev V, Koepsell H, Pastor-Anglada M. 2014. Nucleoside transporters and human organic cation transporter 1 determine the cellular handling of DNA-methyltransferase inhibitors. Br J Pharmacol. 171(16):3868–3880.
- Beumer JH, Eiseman JL, Parise RA, Florian JA Jr., Joseph E, D'Argenio DZ, Parker RS, Kay B, Covey JM, Egorin MJ. 2008a. Plasma pharmacokinetics and oral bioavailability of 3,4,5,6-tetrahydrouridine, a cytidine deaminase inhibitor, in mice. Cancer Chemother Pharmacol. 62(3):457–464.
- Beumer JH, Parise RA, Newman EM, Doroshow JH, Synold TW, Lenz HJ, Egorin MJ. 2008b. Concentrations of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU). Cancer Chemother Pharmacol. 62(2):363–368.
- CDC. 2020. Data & statistics on sickle cell disease; [accessed 2021 Jan 12]. https://www.cdc.gov/ncbddd/sicklecell/data.html
- Charache S, Dover G, Smith K, Talbot CC Jr., Moyer M, Boyer S. 1983. Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta-beta-globin gene complex. Proc Natl Acad Sci USA. 80(15):4842–4846.
- Cohen RM, Wolfenden R. 1971. Cytidine deaminase from Escherichia coli: purification, properties and inhibition by the potential transition state analog 3,4,5,6-tetrahydrouridine. J Biol Chem. 246(24):7561–7565.
- Damaraju VL, Mowles D, Yao S, Ng A, Young JD, Cass CE, Tong Z. 2012. Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids. 31(3):236–255.
- DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, et al. 2019. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 133(1):7–17.
- EMA. 2008. Hydroxycarbamide (Siklos) EPAR – product information; [accessed 2020 Nov 24]. https://www.ema.europa.eu/en/documents/product-information/siklos-epar-product-information_en.pdf
- EMA. 2012a. Dacogen Assessment report (EMA/620205/2012); [accessed 2020 Nov 18]. https://www.ema.europa.eu/en/documents/assessment-report/dacogen-epar-public-assessment-report_en.pdf
- EMA. 2012b. Guideline on the investigation of drug interactions (CPMP/EWP/560/95/Rev. 1 Corr. 2); [accessed 2020 Nov 24]. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf
- EMA. 2017. Public summary of opinion on orphan designation – decitabine and tetrahydrouridine for the treatment of sickle cell disease (EMA/324973/2017); [accessed 2021 Jan 12]. https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/17/1881-public-summary-opinion-orphan-designation-decitabine-tetrahydrouridine-treatment-sickle-cell_en.pdf
- EMA. 2019. Decitabine (Dacogen®): EPAR – product information; [accessed 2021 Jan 13]. https://www.ema.europa.eu/en/documents/product-information/dacogen-epar-product-information_en.pdf
- EMA. 2021. Crizanlizumab (Adakveo) – EPAR product information; [accessed 2021 Jan 12]. https://www.ema.europa.eu/en/documents/product-information/adakveo-epar-product-information_en.pdf
- FDA. 2017. L-glutamine (EndariTM) – prescribing information; [accessed 2020 Nov 24]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208587s000lbl.pdf
- FDA. 2018. Decitabine (DACOGEN®) – prescribing information; [accessed 2021 Jan 13]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021790s021lbl.pdf
- FDA. 2019a. Crizanlizumab-tmca (ADAKVEO®) – prescribing information; [accessed 2021 May 5]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761128s000lbl.pdf
- FDA. 2019b. Venetoclax (VENCLEXTA®) – prescribing information; [accessed 2021 Nov 10]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/208573s013lbl.pdf
- FDA. 2019c. Voxelotor (OXBRYTA™) – prescribing information; [accessed 2020 Nov 24]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/213137s000lbl.pdf
- FDA. 2020. In vitro drug interaction studies – cytochrome P450 enzyme and transporter-mediated drug interactions – guidance for industry; [accessed 2020 Nov 24]. https://www.fda.gov/media/134582/download
- Giacomini KM, Huang S-M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. 2010. Membrane transporters in drug development. Nat Rev Drug Discov. 9(3):215–236.
- Govindarajan R, Bakken AH, Hudkins KL, Lai Y, Casado FJ, Pastor-Anglada M, Tse CM, Hayashi J, Unadkat JD. 2007. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol. 293(5):R1809–R1822.
- Jordheim LP, Durantel D, Zoulim F, Dumontet C. 2013. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 12(6):447–464.
- Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, et al. 2018. Sickle cell disease. Nat Rev Dis Primers. 4:18010.
- Kreis W, Budman DR, Chan K, Allen SL, Schulman P, Lichtman S, Weiselberg L, Schuster M, Freeman J, Akerman S, et al. 1991. Therapy of refractory/relapsed acute leukemia with cytosine arabinoside plus tetrahydrouridine (an inhibitor of cytidine deaminase)–a pilot study. Leukemia. 5(11):991–998.
- Lavelle D, Vaitkus K, Ling Y, Ruiz MA, Mahfouz R, Ng KP, Negrotto S, Smith N, Terse P, Engelke KJ, et al. 2012. Effects of tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral decitabine. Blood. 119(5):1240–1247.
- Liang Y, Li S, Chen L. 2015. The physiological role of drug transporters. Protein Cell. 6(5):334–350.
- Manwani D, Frenette PS. 2013. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. 122(24):3892–3898.
- Marsh JH, Kreis W, Barile B, Akerman S, Schulman P, Allen SL, DeMarco LC, Schuster MW, Budman DR. 1993. Therapy of refractory/relapsed acute myeloid leukemia and blast crisis of chronic myeloid leukemia with the combination of cytosine arabinoside, tetrahydrouridine, and carboplatin. Cancer Chemother Pharmacol. 31(6):481–484.
- Molokie R, Lavelle D, Gowhari M, Pacini M, Krauz L, Hassan J, Ibanez V, Ruiz MA, Ng KP, Woost P, et al. 2017. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Med. 14(9):e1002382.
- Paikari A, Sheehan VA. 2018. Fetal haemoglobin induction in sickle cell disease. Br J Haematol. 180(2):189–200.
- Piel FB, Steinberg MH, Rees DC. 2017. Sickle cell disease. N Engl J Med. 376(16):1561–1573.
- Saunthararajah Y. 2019. Targeting sickle cell disease root-cause pathophysiology with small molecules. Haematologica. 104(9):1720–1730.
- Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. 2014. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 123(4):481–485.
- Varga A, Lionne C, Roy B. 2016. Intracellular metabolism of nucleoside/nucleotide analogues: a bottleneck to reach active drugs on HIV reverse transcriptase. Curr Drug Metab. 17(3):237–252.