
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
For ester prodrugs that are used to improve the gastrointestinal absorption of highly hydrophilic, pharmacologically active substances, it is challenging to predict the human pharmacokinetics (PK) of the prodrugs and their parent compounds using only preclinical data.
This research was aimed at constructing a PBPK model for predicting the human PK of the ester prodrug MGS0274 and its parent compound MGS0008 after a single oral administration of MGS0274 besylate.
First, we identified carboxylesterase 1 (CES1) as the major enzyme involved in the hydrolysis of MGS0274. Second, we constructed a new compartment model to estimate the passive diffusion clearance (CLpd) of MGS0008, a critical parameter for predicting the PK of highly hydrophilic compounds, based on in vivo monkey PK data. Finally, we constructed a permeability-limited liver PBPK model incorporating the CLpd assumed to be the same in humans.
We confirmed that our method reliably predicted the human PK and that the estimated CLpd was comparable to that calculated retrospectively using the PBPK model, suggesting that the methodology for estimating the CLpd was valid.
Our proposed methodology is expected to be helpful for human PK prediction of ester prodrugs hydrolysed by CES1 and their hydrophilic parent compounds even during the preclinical phase.
Introduction
The prodrug strategy is one of the chemical modification approaches for active pharmaceutical substances and is frequently used for improving the gastrointestinal absorption, tissue distribution, and chemical stability of the parent compounds. Indeed, more than 12% of all the small-molecule drugs approved by the US Food and Drug Administration from 2008 to 2017 were prodrugs, and one-third of them were ester prodrugs designed to improve the gastrointestinal absorption of their parent compounds (Rautio et al. Citation2018). While the prodrug strategy is one of the effective strategies to overcome some pharmacokinetic problems of the parent compounds, it also poses challenges, because, as compared to the case for non-prodrugs, the human pharmacokinetics (PK) of prodrugs and their parent compounds are not easy to predict during the preclinical phase of drug development (Rautio et al. Citation2018). Therefore, one of the important challenges to increase the success rate of prodrug development is to construct an accurate human PK prediction method for prodrugs and their parent compounds based only on preclinical data.
The physiologically based pharmacokinetic (PBPK) modelling is a powerful tool, not only for predicting the PK of compounds in the plasma and target tissues, but also for predicting potential drug-drug interactions; therefore, it is widely used in various phases of drug development (Jones et al. Citation2015). Although PBPK models have been used frequently for predicting the human PK of compounds that are primarily metabolised by cytochrome P450 enzymes (Sager et al. Citation2015), they have been scarcely used for compounds that are metabolised by other enzymes such as esterases, which frequently metabolise ester prodrugs. To date, efforts to predict the human PK of several ester prodrugs have been reported; e.g., oseltamivir, valganciclovir, and mycophenolate mofetil (Parrott et al. Citation2011; Malmborg and Ploeger Citation2013; Lukacova et al. Citation2016). For the cases of oseltamivir and valganciclovir, their human PK were simulated using not only preclinical data but also actual human PK data before clinical trials to assess drug-drug interactions and paediatric studies were initiated. For mycophenolate mofetil, PBPK modelling was introduced as a screening tool in the early phases of drug development, using only preclinical data obtained from in vitro assays and studies on physicochemical properties; however, the authors also mentioned the need for further improvements before candidate selection for phase 1 clinical trials (Malmborg and Ploeger Citation2013). Thus, the methods described above would not be suitable for accurate prediction of the human PK of ester prodrugs and their parent compounds in the preclinical phase.
Species differences are known to exist in the activities (Nishimuta et al. Citation2014) and tissue distribution of esterases (Di Citation2019); therefore, identification of the esterase isoforms involved in the hydrolysis of ester prodrugs is essential for accurate prediction of the human PK of ester prodrugs and their parent compounds. Moreover, since the parent compounds are generally more polar and less membrane-permeable than their ester prodrugs, their expected low membrane permeability would be a key property for accurate human PK prediction (Hu et al. Citation2014; Lukacova et al. Citation2016). However, no PBPK models based on only preclinical data that took the above two points into consideration have been reported until date.
This research was aimed at constructing a PBPK model based on only preclinical data for accurate human PK prediction of an ester prodrug MGS0274 and its hydrophilic parent compound MGS0008, a metabotropic glutamate 2/3 receptor agonist (Kinoshita et al. Citation2019; Urabe et al. Citation2020;), after a single oral administration of MGS0274 besylate, which is under clinical development for the treatment of schizophrenia (development code: TS-134). MGS0274 besylate has shown to successfully improve the oral bioavailability of MGS0008 by about 20-fold in monkeys (Kinoshita et al. Citation2019), and to be almost completely and presystemically hydrolysed into MGS0008 in humans (Watanabe et al. Citation2020). First, we conducted esterase-phenotyping studies using several esterase inhibitors and recombinant enzymes and identified carboxylesterase 1 (CES1) as the enzyme primarily involved in the hydrolysis of MGS0274. Second, we constructed a new compartment model to estimate the passive diffusion clearance (CLpd) of MGS0008 using the in vivo PK data in monkeys, for which the amino acid sequences and tissue distribution patterns of CES1 are close to those in humans among experimental animals (Di Citation2019). Finally, we constructed a permeability-limited liver PBPK model using the Simcyp simulator (Version 18; Simcyp Ltd., Sheffield, UK), incorporating the CLpd in monkeys which was assumed to be the same in humans, and successfully predicted the human PK for both MGS0274 and MGS0008 after oral dosing of the prodrug. The proposed methodology is expected to be helpful for human PK prediction of ester prodrugs, which are hydrolysed by CES1 and releasing highly polar parent compounds, even in the preclinical phase of drug development.
Materials and methods
Materials
MGS0274 besylate, MGS0008, [2H4]MGS0274 besylate, and [13C215N]MGS0008 were synthesised at Taisho Pharmaceutical Co., Ltd. (Saitama, Japan). Benzil, bis(p-nitrophenyl)phosphate (BNPP), and p-chloromercuribenzoate (PCMB) were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Ethephon and phenylmethane sulphonyl fluoride (PMSF) were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan) and Honeywell International Inc. (Charlotte, NC, USA), respectively. All other reagents were of analytical grade or of high-performance liquid chromatography grade. Human liver S9 fractions (mixed gender) were purchased from Sekisui XenoTech, LLC (Kansas City, KS, USA). Recombinant human CES1 and CES2 bactosomes were purchased from Cypex Ltd. (Dundee, UK).
Parallel artificial membrane permeability assay (PAMPA)
The membrane permeability of MGS0274 was measured using a PAMPA Evolution instrument (Pion Inc., Billerica, MA, USA). A STIRWELL PAMPA Sandwich plate consisting of a donor bottom plate and an acceptor filter plate was used for the experiment. The donor wells were filled with donor solutions prepared with PRISMA HT adjusted to pH 6.2, which contained MGS0274 besylate (final concentration of 10 μM). An acceptor well was coated with GIT-0 Lipid Solution, followed by the addition of Acceptor Sink Buffer (pH 7.4). After incubation for 4 hours at room temperature, aliquots of the donor and acceptor solutions were separately collected and mixed with appropriate volumes of the corresponding blank buffer to match the matrix, and acetonitrile/methanol/formic acid (90:10:1, v/v/v) containing [2H4]MGS0274 was added as an internal standard (IS). The resultant mixtures were centrifuged at 3,974 ×g for 10 minutes at 4 °C, and aliquots of the supernatants were subjected to bioanalysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The apparent permeability (Papp) was calculated using the PAMPA Evolution software (version 3.8; Pion Inc.).
Blood-to-plasma ratio
The blood-to-plasma ratios of MGS0274 and MGS0008 were analysed using pooled fresh whole blood and control plasma separated from fresh whole blood from humans. The anticoagulants used for MGS0274 and MGS0008 were sodium heparin and dipotassium ethylene diamine tetraacetate, respectively. Whole blood and control plasma containing 2.5 or 250 nM of MGS0274 besylate or 3 μM of MGS0008 were incubated at 37 °C for 30 minutes in a shaking water bath. The incubated whole blood was centrifuged at 2,000 ×g for 10 minutes at 4 °C to obtain plasma samples. An aliquot of each plasma sample (that is, isolated plasma from incubated whole blood and control plasma) was mixed with eight volumes of acetonitrile/methanol/formic acid (90:10:1, v/v/v) containing [2H4]MGS0274 and [13C215N]MGS0008 as each IS for bioanalysis. After centrifugation at 3,974 ×g for 10 minutes at 4 °C, the supernatant was subjected to bioanalysis by LC-MS/MS. The blood-to-plasma ratio was calculated by dividing the concentration in the control plasma, which was the same as that in whole blood, by that in the plasma obtained by centrifugation from the incubated whole blood.
Enzyme kinetics for hydrolysis of MGS0274 into MGS0008 in human liver S9 fractions
Human liver S9 fractions with a protein concentration of 0.25 mg/mL in sodium potassium phosphate buffer (pH 7.4) consisting of sodium phosphate (250 mM), KCl (75 mM), MgCl2 (2.4 mM), glucose-6-phosphate (1.5 mM), and NADP+ (0.16 mM) were pre-incubated for 5 minutes at 37 °C, and the reactions were initiated by the addition of methanol solution of MGS0274 besylate at the final concentrations of 1 to 1000 μM. The final concentration of organic solvent in the incubation mixture was 1.2% (v/v) or less. After incubation for 1, 2, or 5 minutes at 37 °C, the reactions were terminated by the addition of two volumes of acetonitrile/methanol/formic acid (90:10:1, v/v/v) containing [13C215N]MGS0008. The resultant mixtures were centrifuged at 2,150 ×g for 10 minutes at 4 °C, and aliquots of the supernatants were subjected to bioanalysis by LC-MS/MS.
The formation rates (nmol/min/mg protein) of MGS0008 in the reaction mixtures were calculated by EquationEquation (1)(1)
(1) :
(1)
(1)
The kinetic parameters were calculated using Phoenix WinNonlin (version 8.0; Certara, Princeton, NJ, USA) using EquationEquation (2)(2)
(2) , a nonlinear least-squares regression fitting to the modified two-site Michaelis-Menten equation:
(2)
(2)
where CLint is the intrinsic clearance derived from low-affinity enzymes, Km is the kinetic constant, S is the concentration of MGS0274 besylate, V is the formation rate (initial velocity), and Vmax is the maximum velocity.
Esterase reaction phenotyping for hydrolysis of MGS0274 into MGS0008 in human liver S9 fractions
Human liver S9 fractions with a protein concentration of 0.625 mg/mL in homogenisation buffer (pH 7.4) consisting of sodium phosphate (10 mM) and KCl (150 mM) were kept on ice for 30 minutes with benzil, BNPP, ethephon, PCMB, or PMSF dissolved in dimethyl sulfoxide, the final concentration of which was 0.25, 2.5, 0.25, 0.25, or 2.5 mM, respectively. Each mixture (100 μL) was mixed with a co-factor solution (150 μL) consisting of sodium phosphate (416 mM), KCl (25 mM), MgCl2 (4.1 mM), glucose-6-phosphate (2.4 mM), and NADP+ (0.26 mM), and then pre-incubated for 5 minutes at 37 °C. The reactions were initiated by the addition of a methanol solution of MGS0274 besylate at the final concentration of 10 μM. The final concentration of organic solvent in the incubation mixture was 1.7% (v/v) or less. After incubation for 5 minutes at 37 °C, the reactions were terminated by the addition of two volumes of acetonitrile/methanol/formic acid (90:10:1, v/v/v) containing [13C215N]MGS0008. The resultant mixtures were centrifuged at 2,150 ×g for 10 minutes at 4 °C, and aliquots of the supernatants were subjected to bioanalysis by LC-MS/MS. The percent inhibition was calculated by EquationEquation (3)(3)
(3) .
(3)
(3)
where Ccontrol is the concentration of MGS0008 in the absence of inhibitors and Cinhibited is the concentration of MGS0008 in the presence of inhibitors.
Carboxylesterase reaction phenotyping for hydrolysis of MGS0274 into MGS0008
Recombinant human CES1 or CES2 bactosomes with a protein concentration of 75 μg/mL in phosphate buffer (49 mM, pH 7.4) were pre-incubated for 5 minutes at 37 °C, and the reactions were initiated by the addition of a methanol solution of MGS0274 besylate at the final concentration of 10 μM. The final concentration of organic solvent in the incubation mixture was 2.0% (v/v). After incubation for 5, 15, 30, or 60 minutes, the reactions were terminated by the addition of two volumes of acetonitrile/methanol/formic acid (90:10:1, v/v/v) containing [13C215N]MGS0008. The resultant mixtures were centrifuged at 2,150 ×g for 10 minutes at 4 °C, and aliquots of the supernatants were subjected to bioanalysis by LC-MS/MS. The formation rates of MGS0008 in the reaction mixtures were calculated using EquationEquation (1)(1)
(1) .
LC-MS/MS conditions
For MGS0008, the HPLC system consisted of an LC-10AD, LC-20AD, or LC-30AD system (Shimadzu, Kyoto, Japan). Chromatographic separation was performed using an Xbridge Amide column (3.5 μm, 50 mm × 4.6 mm I.D.; Waters, Milford, MA, USA) with 0.1% (v/v) formic acid (A) and acetonitrile (B) as the mobile phase under a gradient condition at a flow rate of 1.0 mL/min. A linear gradient was applied as follows: 0.00–0.30 minutes, hold isocratic at 90% B; 0.30–2.50 minutes, from 90 to 50% B; 2.50–3.00 minutes, hold isocratic at 50% B; 3.00–3.01 minutes, from 50% to 90% B; 3.01–4.00 minutes, hold isocratic at 90% B. The temperatures of the column and the sample compartment were set to 50 and 15 °C, respectively. The sample measurement was performed using API4000TM or TripleQuadTM 5500 (AB Sciex, Framingham, MA, USA) with the TurboIonSpray ionisation mode in the positive ion detection mode for MGS0008 (m/z 204 → 158) and [13C215N]MGS0008 (m/z 207 → 160). For MGS0274, the HPLC system consisted of an LC-20AD or LC-30AD system. Chromatographic separation was performed using a Shim-pack XR-ODS (2.2 μm, 30 mm × 3.0 mm I.D.; Shimadzu) with 0.1% (v/v) formic acid (A) and acetonitrile (B) as the mobile phase under a gradient condition at a flow rate of 1.3 mL/min. A linear gradient was applied as follows: 0.00–1.00 minutes, from 2 to 98% B; 1.00–1.20 minutes, hold isocratic at 98% B; 1.20–1.21 minutes, from 98 to 2% B; 1.21–1.50 minutes, hold isocratic at 2% B. The temperatures of the column and the sample compartment were set to 50 and 10 °C, respectively. The sample measurement was performed using API4000TM or TripleQuadTM 5500 with the TurboIonSpray ionisation mode in the positive ion detection mode for MGS0274 (m/z 430 → 386) and [2H4]MGS0274 (m/z 434 → 390).
Estimation of the passive diffusion clearance of MGS0008 for monkeys
A compartment model analysis using Phoenix WinNonlin (version 8.0) was conducted to estimate the CLpd of MGS0008 from the PK data following an oral administration of MGS0274 besylate to male cynomolgus monkeys at the dose of 2.89 mg/kg (1 mg eq. of MGS0008/kg), which were reported from a previous study (Kinoshita et al. Citation2019). No carrier-mediated transport across the membrane was assumed in this model, since it was not always easy to examine this in the preclinical phase of drug development, due to the lack of sufficient information to design an appropriate experiment. Mass balance equations were described with the molar concentrations, as MGS0274 was hydrolysed into equimolar amounts of MGS0008 (). The compartment model was designed as one in which MGS0274 was immediately and completely hydrolysed into metabolically stable MGS0008 in the intestine and/or the liver, based on the results of our previous in vitro and in vivo metabolism studies (Kinoshita et al. Citation2019). The contributions of hydrolysis in the intestine and the liver were determined by the intestinal availability (Fg) value, which was calculated using the Qgut model (Yang et al. Citation2007) based on the MGS0274 hydrolytic activity in monkey intestine S9 fractions (Kinoshita et al. Citation2019). The CLpd was calculated by multiplying the specific cellular permeation rate constant (kspec) by the tissue cell volume (Lukacova et al. Citation2016) with reference to the parameter of the permeability and surface area product, which is commonly used to describe the rate of passive diffusion. In addition, the contribution of the basolateral cell surface in tissues needs to be considered for accurate calculation of the CLpd. In this study, the contribution of the basolateral cell surface in the intestine and the liver was set to be a half of and two thirds of the total cell surface, respectively, based on the literature (Trapa et al. Citation2017; Blouin Citation1977). The kspec of MGS0008 was estimated from the observed monkey PK data using the maximum likelihood estimation, and the CLpd of MGS0008 for the intestine was obtained using EquationEquation (5)(5)
(5) . Then, the CLpd of MGS0008 for the liver was calculated by EquationEquation (8)
(8)
(8) with the estimated kspec and converted to that for hepatocytes using EquationEquation (10)
(10)
(10) . Note that the fraction absorbed (Fa) was defined as the oral bioavailability of MGS0008 after a single oral administration of MGS0274 besylate, so that presystemic excretion of the compounds out of the body, such as the luminal and bile secretion through first-pass, was also incorporated.
The compartment model scheme and input parameters are shown in and , respectively. The mass balance equations are shown below:
Intestine compartment
(4)
Liver compartment
Systemic plasma compartment
Table 1. Input parameters used in the pharmacokinetic compartment models for monkeys.
PBPK modelling and simulation
Human PBPK models were developed using the Simcyp simulator (Version 18) and the input parameters are summarised in . The steady-state volumes of distribution (Vdss) of MGS0274 and MGS0008 were predicted by a monkey single-species allometric scaling method (SSSmonkey) (Lombardo et al. Citation2013). The distribution of MGS0274 and MGS0008 were predicted using the full PBPK model and tissue-to-plasma partition method proposed by Rodgers and Rowland (Method 2 in Simcyp; Rodgers and Rowland Citation2007). The Vdss calculated by Method 2 was adjusted using tissue-to-plasma partition coefficient (Kp) scalar to match the predicted Vdss from SSSmonkey. Since MGS0008 would have low membrane permeability due to its high hydrophilicity, the permeability-limited liver model with the CLpd for hepatocytes was applied to depict the gradual increase in its plasma concentrations, as in monkeys. The Km and Vmax in human liver S9 fractions were inputted as the kinetic parameters of the enzyme which was specified from the results of the esterase-phenotyping studies to predict the in vivo clearance of MGS0274 based on the correlation between in vitro CLint and in vivo CLint for CES1 substrates (Nishimuta et al. Citation2014). The human CLtotal of MGS0008 predicted from SSSmonkey was set at the renal clearance, based on the assumption that urinary clearance was the main route of elimination of MGS0008 in humans, as in rats and monkeys (Kinoshita et al. Citation2019). The plasma concentration-time profiles following a single oral administration of MGS0274 besylate under the fed condition were simulated in 10 virtual trials of 10 healthy volunteers (age: 20–50 years old, gender ratio: 0.5) over the dose range of 10–100 mg as the free base, in which the built-in ‘Sim-Healthy Volunteers’ population was used without modifications.
Table 2. Input parameters in the PBPK models for humans.
Evaluation of the prediction accuracy
The fold error (FE) of the difference between the predicted and observed PK parameters in humans (Watanabe et al. Citation2020) were calculated by dividing the predicted value by the observed value.
Results
PAMPA and the blood-to-plasma ratio
The Papp of MGS0274 in PAMPA at pH 6.2 was 13.0 × 10−6 cm/s. The blood-to-plasma ratio of MGS0274 was 0.690 at 2.5 nM and 0.631 at 250 nM, and that of MGS0008 was 0.555 at 3 μM.
Enzyme kinetics for hydrolysis of MGS0274 into MGS0008 in human liver S9 fractions
The formation rates of MGS0008 ranged from 0.351 to 16.0 nmol/min/mg protein over the entire concentration range tested of MGS0274. The Eadie–Hofstee plot was found to be curvilinear, indicating the contribution of two enzymes (Clarke Citation1998). The Km and Vmax for the high-affinity enzyme in human liver S9 fractions were calculated to be 17.5 μM and 8.87 nmol/min/mg protein, respectively. The CLint for the low-affinity enzyme was calculated to be 0.00529 mL/min/mg protein, which was about 100-fold lower than the CLint simply calculated by dividing the Vmax by Km (0.507 mL/min/mg protein), suggesting that most of the MGS0274 hydrolytic activity in human liver S9 fractions was derived from the high-affinity enzyme.
Esterase reaction phenotyping for hydrolysis of MGS0274 into MGS0008 in human liver S9 fractions
The inhibitory effects of several esterase inhibitors against MGS0274 hydrolytic activity in human liver S9 fractions were evaluated, and the percent inhibitions are shown in . Formation of MGS0008 was not inhibited by ethephon or PCMB, indicating that butyrylcholinesterase, carboxymethylenebutenolidase, and paraoxonase were not involved in the hydrolysis of MGS0274. On the other hand, both BNPP and PMSF completely inhibited the formation of MGS0008, indicating that arylacetamide deacetylase (AADAC) and/or CES were involved in the hydrolysis of MGS0274. In addition, the formation of MGS0008 was also inhibited completely by benzil, which had little inhibitory effect against AADAC activity (Oda et al. Citation2015), suggesting that some CES isoforms are involved in the hydrolysis of MGS0274.
Table 3. Characteristic and inhibitory effects of esterase inhibitors on the hydrolysis of MGS0274 in human liver S9 fractions.
Carboxylesterase reaction phenotyping for hydrolysis of MGS0274 into MGS0008
The formation rate of MGS0008 in recombinant human CES1 bactosomes was 0.918 nmol/min/mg protein, which was much higher than that in recombinant human CES2 bactosomes (0.0344 nmol/min/mg protein), suggesting that CES1 was mainly responsible for the enzymatic hydrolysis of MGS0274 into MGS0008.
Estimation of the passive diffusion clearance of MGS0008 for monkeys
Using the model inputs detailed in , the kspec was obtained by fitting the observed monkey PK data of MGS0008 obtained following a single oral administration of MGS0274 besylate with the compartment model, shown in , and the CLpd,i and CLpd,l for monkeys were calculated to be 0.144 and 5.45 mL/h/kg, respectively. The obtained CLpd,l was converted to the CLpd for hepatocytes of 2.37 × 10−5 mL/min/million cells.
Simulation of human pharmacokinetic profiles using PBPK model
Using the model inputs detailed in , the plasma exposures to MGS0274 and MGS0008 in humans after a single oral administration of MGS0274 besylate under the fed condition were simulated for the dose range of 10–100 mg as the free base, and their increases in a dose-dependent manner were confirmed. The simulated plasma concentration-time profiles of MGS0274 and MGS0008 and their PK parameters at the dose of 10 mg as free base, at which food effects on the pharmacokinetics was evaluated in the phase 1 study (Watanabe et al. Citation2020), are shown in and , respectively. The simulated plasma concentration of MGS0274 rapidly reached a maximum concentration (Cmax) of 13.88 ng/mL at 0.37 hours, and declined at a terminal half-life (t1/2) of 2.272 hours, and that of MGS0008 reached a Cmax of 45.21 ng/mL at 3.9 hours and declined at a t1/2 of 3.870 hours. The area under the concentration-time curve from zero to infinity (AUC0–inf) of MGS0274 and MGS0008 were 31.96 h*ng/mL and 463.5 h*ng/mL, respectively, and the AUC0–inf of MGS0274 was no more than 3% against that of MGS0008 in a molar concentration.
Table 4. Predicted and observed pharmacokinetic parameters of MGS0274 and MGS0008 in humans after a single oral administration of MGS0274 besylate (10 mg as MGS0274) under the fed condition.
Evaluation of the prediction accuracy
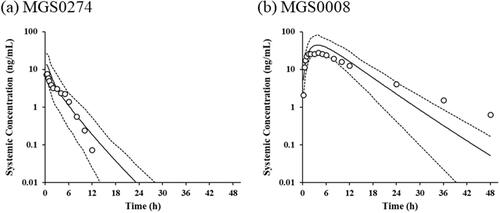
The simulated plasma concentration-time profiles and predicted PK parameters, along with the observed data at the dose of 10 mg as free base of MGS0274 besylate in healthy volunteers reported in the literature (Watanabe et al. Citation2020) are shown in and , respectively. The observed mean plasma concentrations of MGS0274 and MGS0008 after dosing were within the 5th and 95th percentile of the simulated plasma concentrations, except for those of MGS0008 at 36 and 48 hours, which were affected by the difference in the t1/2 of MGS0008 (observed value: 8.699 hours; predicted value: 3.870 hours). The predicted mean tmax of MGS0008 (3.9 hours) was approximately equal to the observed value (4.0 hours), which corresponds to the characteristics of a highly hydrophilic compound like MGS0008. The FE of Cmax and AUC0–inf were 1.7 and 1.4 for MGS0274, and 1.6 and 1.2 for MGS0008, respectively.
Discussion
PBPK modelling is frequently used for predicting the human PK of new drug candidates. For ester prodrugs which are designed to improve the gastrointestinal absorption of their hydrophilic parent compounds, the CLpd is a critical parameter for successful PBPK modelling (Hu et al. Citation2014; Lukacova et al. Citation2016). Herein, we have proposed a new methodology for accurately predicting the human PK of the ester prodrug MGS0274 and its parent compound MGS0008 after a single oral administration of MGS0274 besylate, based on only preclinical data obtained prior to the initiation of the phase 1 clinical trial. This study consisted of three steps: identification of the enzymes and tissues involved in the activation of the prodrug in humans, estimation of the CLpd of MGS0008 in animals based on the compartmental analysis of the in vivo animal PK data, and construction of a permeability-limited PBPK model incorporating the CLpd in animals which was assumed to be the same in humans for predicting the human PK.
Identification of the enzymes responsible for the activation of prodrugs and of their tissue distribution is essential for accurate prediction of the human PK of the parent compounds. The esterase reaction phenotyping assay conducted using human liver S9 fractions and the subsequent carboxylesterase reaction phenotyping assay with recombinant human CESs revealed that CES1 was primarily responsible for the hydrolysis of MGS0274 into MGS0008. Actually, MGS0274 shares some structural characteristics with CES1 substrates (Fukami and Yokoi Citation2012), including the large acyl group (MGS0008) and small alcohol group (l-menthol as a side-chain moiety of the prodrug). Furthermore, our previous study revealed high MGS0274 hydrolytic activity in tissues in which CES1 was strongly expressed, such as rat serum, monkey intestine and liver, and human liver (Kinoshita et al. Citation2019). In humans, therefore, MGS0274 would primarily be hydrolysed into MGS0008 in the liver, and not the intestine.
We have already reported that the plasma t1/2 of MGS0008 in monkeys after a single oral administration of MGS0274 besylate was approximately 10-fold more prolonged as compared with that after a single intravenous administration of MGS0008, which would be explained by the rate-limiting release of MGS0008 from the enterocytes and/or hepatocytes into the plasma (Kinoshita et al. Citation2019). As the main esterase involved in MGS0274 activation is CES1, which is abundantly expressed in the human liver, the CLpd of MGS0008 for hepatocytes should be a critical parameter for accurately predicting the human PK, as the release is rate-limiting. Some methods have been reported so far, in which the CLpd is estimated based on hepatic uptake assays in the presence of transporter inhibitors or at low temperatures (Bi et al. Citation2017), or based on the relationship between the Log D7.4 values and passive permeability of reference compounds across the hepatocyte membrane in the presence of transporter inhibitors (De Bruyn et al. Citation2018). However, in the case of use of inhibitors or low temperatures, it would be difficult to avoid substrate-dependent inhibition or alterations of the membrane fluidity, which may affect the results (Li et al. Citation2014). Estimation of the CLpd for the liver from the lipophilicity was also deemed as being inappropriate to apply to MGS0008, of which cLog D7.4 value of −4.18 (ACD/percepta version 14.50; Advanced Chemistry Development, Toronto, Canada) fell outside the range for the reference compounds (Log D7.4: −1.1–2.5) reported in the literature (De Bruyn et al. Citation2018). In fact, when the CLpd estimated from the cLog D7.4 value of MGS0008 was used, the simulated human plasma concentration-time profiles of MGS0008 were much lower and sustained than clinically observed values, suggesting that the estimated CLpd was significantly lower than the true value (Supplemental Figure 1 (a)). Thus, for the reasons mentioned above, we decided to develop a new compartment model to estimate the CLpd for the human liver from the actual in vivo animal PK data of MGS0008 obtained after oral administration of MGS0274 besylate.
Figure 1. Bioactivation of MGS0274 MGS0274 undergoes spontaneous enzymatic hydrolysis into MGS0008, l-menthol, acetaldehyde, and carbon dioxide. No other metabolites have been reported from either in vitro studies (rat, monkey, and human hepatocytes) or in vivo studies (rats and monkeys) (Kinoshita et al. Citation2019).
Figure 2. Pharmacokinetic compartment model scheme. Cintestine: concentration of MGS0008 in the intestine; Cliver: concentration of MGS0008 in the liver; Cportal,274: plasma concentration of MGS0274 in the portal vein; Csys: systemic plasma concentration of MGS0008; CLmet,274: metabolic clearance from MGS0274 into MGS0008; CLpd,i: passive diffusion clearance of MGS0008 for the intestine; CLpd,l: passive diffusion clearance of MGS0008 for the liver; CLtotal: total clearance of MGS0008.
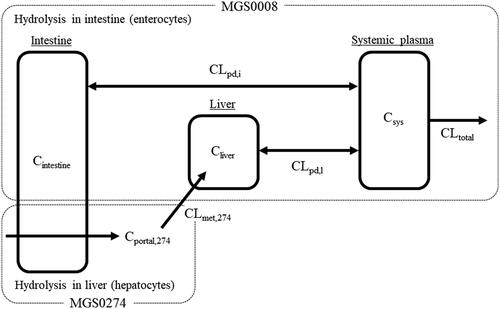
Figure 3. PBPK-simulated plasma concentration-time profiles and the observed plasma concentrations of MGS0274 and MGS0008 in humans after a single oral administration of MGS0274 besylate (10 mg as MGS0274) under the fed condition The solid lines show the PBPK-simulated mean plasma concentration-time profiles of (a) MGS0274 and (b) MGS0008. The dotted lines represent the 5th and 95th percentiles. Open circles represent the mean plasma concentrations of MGS0274 and MGS0008 obtained in the single-ascending dose study, the clinical trial design of which is reported in the literature (Watanabe et al. Citation2020). PBPK: physiologically based pharmacokinetics.
Among the experimental animals used during the processes of drug development, monkeys are often selected for evaluating the in vivo PK of the substrates of CES1, because the amino acid sequences and tissue distribution patterns of CES1 are close to those in humans, although intestinal expression of CES1 is observed in monkeys (Di Citation2019). In fact, the MGS0274 hydrolytic activity in the monkey intestinal S9 fractions was as high as that in the monkey liver S9 fractions (Kinoshita et al. Citation2019), and the calculated Fg of MGS0274 was low (0.06), suggesting that almost all MGS0274 would be hydrolysed during intestinal absorption in monkeys. Accordingly, there was a need to develop a new compartment model that would enable both estimation of the CLpd for the intestine and its conversion to the CLpd for the liver. Based on the assumption that the parameter representing the kspec of MGS0008 was not organ- or species-specific, but rather compound-specific (Lukacova et al. Citation2016), the CLpd for both the intestine and the liver were described by the kspec in the compartment model. In addition, the intestine was defined as one-compartment of duodenum since MGS0274 was highly lipophilic and highly permeable compound which would be absorbed mainly in the upper intestine, duodenum (Masaoka et al. Citation2006). With this compartment model, the kspec of MGS0008 was estimated from the observed monkey PK data, and the CLpd of MGS0008 for the intestine was determined. Then, the CLpd of MGS0008 for the liver was calculated using the estimated kspec and converted to that for hepatocytes based on some physiological parameters, such as the hepatocellularity and the liver weight (Hosea et al. Citation2009). The CLpd for human hepatocytes is assumed to be equal to that for monkey hepatocytes, since similarity of the CLpd between human and monkey hepatocytes has been reported (De Bruyn et al. Citation2018). Note that use of portal vein-cannulated monkeys might be more useful to obtain the CLpd of MGS0008 for the intestine than the compartment model; however, it is not easy to conduct such studies, and challenges have still remained in terms of how species- and organ-differences of the CLpd can be overcome/corrected for.
The PBPK model, which incorporated a permeability-limited liver module with application of the CLpd for hepatocytes, was constructed using the Simcyp simulator to simulate the human plasma concentration-time profiles of MGS0274 and MGS0008 after a single oral administration of MGS0274 besylate under the fed condition. With this model, a linear increase in the plasma exposure to MGS0008 was predicted over the dose range simulated (10–100 mg as MGS0274), with minimal exposures to MGS0274, corresponding to an AUC0–inf of approximately 3% as compared to that of MGS0008 in a molar concentration, the data being comparable to those observed in the phase 1 clinical trial (Watanabe et al. Citation2020). The predicted Cmax and AUC0–inf of both MGS0274 and MGS0008 at the 10-mg dose were also within 2-fold of the observed values reported in the literature (Watanabe et al. Citation2020), with most of the observed data being within the 5th and 95th percentile range of the simulations. Furthermore, the CLpd of MGS0008 for hepatocytes was calculated retrospectively from the observed human PK data by 1) a sensitivity analysis of the CLpd using the permeability-limited liver PBPK model (0.944 × 10−5 mL/min/million cells), and 2) a compartmental analysis using our compartment model with human physiological and predicted PK parameters listed in and the assumption of no intestinal metabolism of MGS0274 (Fg = 1) (1.69 × 10−5 mL/min/million cells). These CLpd were comparable to the estimated value (2.37 × 10−5 mL/min/million cells). Collectively, these results demonstrated that our methodology to estimate the CLpd of MGS0008 in humans based on the compartmental analysis of the in vivo monkey PK data was valid. It should be noted that when the PBPK model was applied without incorporation of the permeability-limited liver module, the predicated tmax and Cmax of MGS0008 were faster and approximately 4-fold higher than the observed data, respectively (Supplemental Figure 1 (c)). Strictly, the estimated value of CLpd was slightly greater than the retrospectively calculated values, which might have affected the difference in the t1/2 of MGS0008 between the observed (8.699 hours) and predicted (3.870 hours) values. In both the simulated and observed results, MGS0274 was detected in human plasma, as opposed to the case in monkeys; this could be attributed to the species differences in the CES1 expression patterns (CES1 is not expressed in the human intestine).
Conclusion
We proposed a methodology for predicting the human PK of ester prodrugs based on only preclinical data, using a combination of the newly developed compartment model for estimation of the CLpd and the permeability-limited liver PBPK model. Although further validations might be needed for drugs that are hydrolysed incompletely through first-pass hydrolysis or hydrolysed in both the intestine and the liver, our proposed approach is expected to be useful for accurate prediction of the human PK of ester prodrugs even in the preclinical phase of drug development, particularly those hydrolysed by CES1 and releasing highly polar parent compounds.
Supplemental Material
Download PDF (26 KB)Acknowledgments
The authors wish to sincerely thank Shoko Inatani for her support with the data analysis and model construction.
Disclosure statement
The authors report no declarations of interest.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Bi Y, Scialis RJ, Lazzaro S, Mathialagan S, Kimoto E, Keefer J, Zhang H, Vildhede AM, Costales C, Rodrigues AD, et al. 2017. Reliable rate measurements for active and passive hepatic uptake using plated human hepatocytes. AAPS J. 19(3):787–796.
- Blouin A, Bolender RP, Weibel ER. 1977. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. 72(2):441–455.
- Clarke SE. 1998. In vitro assessment of human cytochrome P450. Xenobiotica. 28(12):1167–1202.
- Davies B, Morris T. 1993. Physiological parameters in laboratory animals and humans. Pharm Res. 10(7):1093–1095.
- De Bruyn T, Ufuk A, Cantrill C, Kosa RE, Bi Y, Niosi M, Modi S, Rodrigues AD, Tremaine LM, Varma MVS, et al. 2018. Predicting human clearance of organic anion transporting polypeptide substrates using cynomolgus monkey: in vitro-in vivo scaling of hepatic uptake clearance. Drug Metab Dispos. 46(7):989–1000.
- Di L. 2019. The impact of carboxylesterases in drug metabolism and pharmacokinetics. Curr Drug Metab. 20(2):91–102.
- Fukami T, Yokoi T. 2012. The emerging role of human esterases. Drug Metab Pharmacokinet. 27(5):466–477.
- Hosea NA, Collard WT, Cole S, Maurer TS, Fang RX, Jones H, Kakar SM, Nakai Y, Smith BJ, Webster R, et al. 2009. Prediction of human pharmacokinetics from preclinical information: comparative accuracy of quantitative prediction approaches. J Clin Pharmacol. 49(5):513–533.
- Hu Z, Edginton AN, Laizure SC, Parker RB. 2014. Physiologically based pharmacokinetic modeling of impaired carboxylesterase-1 activity: effects on oseltamivir disposition. Clin Pharmacokinet. 53(9):825–836.
- Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, Snoeys J, Upreti VV, Zheng M, Hall SD. 2015. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 97(3):247–262.
- Kinoshita K, Ochi M, Iwata K, Fukasawa M, Yamaguchi J. 2019. Preclinical disposition of MGS0274 besylate, a prodrug of a potent group II metabotropic glutamate receptor agonist MGS0008 for the treatment of schizophrenia. Pharmacol Res Perspect. 7(5):e00520.
- Li R, Bi Y, Lai Y, Sugano K, Steyn SJ, Trapa PE, Di L. 2014. Permeability comparison between hepatocyte and low efflux MDCKII cell monolayer. AAPS J. 16(4):802–809.
- Lombardo F, Waters NJ, Argikar UA, Dennehy MK, Zhan J, Gunduz M, Harriman SP, Berellini G, Rajlic IL, Obach RS. 2013. Comprehensive assessment of human pharmacokinetic prediction based on in vivo animal pharmacokinetic data, part 1: volume of distribution at steady state. J Clin Pharmacol. 53(2):167–177.
- Lukacova V, Goelzer P, Reddy M, Greig G, Reigner B, Parrott N. 2016. A physiologically based pharmacokinetic model for ganciclovir and its prodrug valganciclovir in adults and children. AAPS J. 18(6):1453–1463.
- Malmborg J, Ploeger BA. 2013. Predicting human exposure of active drug after oral prodrug administration, using a joined in vitro/in silico-in vivo extrapolation and physiologically-based pharmacokinetic modeling approach. J Pharmacol Toxicol Methods. 67(3):203–213.
- Masaoka Y, Tanaka Y, Kataoka M, Sakuma S, Yamashita S. 2006. Site of drug absorption after oral administration: assessment of membrane permeability and luminal concentration of drugs in each segment of gastrointestinal tract. Eur J Pharm Sci. 29(3-4):240–250.
- Nishimuta H, Houston JB, Galetin A. 2014. Hepatic, intestinal, renal, and plasma hydrolysis of prodrugs in human, cynomolgus monkey, dog, and rat: implications for in vitro-in vivo extrapolation of clearance of prodrugs. Drug Metab Dispos. 42(9):1522–1531.
- Oda S, Fukami T, Yokoi T, Nakajima M. 2015. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab Pharmacokinet. 30(1):30–51.
- Parrott N, Davies B, Hoffmann G, Koerner A, Lave T, Prinssen E, Theogaraj E, Singer T. 2011. Development of a physiologically based model for oseltamivir and simulation of pharmacokinetics in neonates and infants. Clin Pharmacokinet. 50(9):613–623.
- Rautio J, Meanwell NA, Di L, Hageman MJ. 2018. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 17(8):559–587.
- Rodgers T, Rowland M. 2007. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 24(5):918–933.
- Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. 2015. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos. 43(11):1823–1837.
- Trapa PE, Beaumont K, Atkinson K, Eng H, King-Ahmad A, Scott DO, Maurer TS, Di L. 2017. In vitro-in vivo extrapolation of intestinal availability for carboxylesterase substrates using portal vein-cannulated monkey. J Pharm Sci. 106(3):898–905.
- Urabe H, Miyakoshi N, Ohtake N, Nozoe A, Ochi M, Fukasawa M, Kinoshita K, Yamaguchi J-I, Marumo T, Hikichi H, et al. 2020. Discovery of MGS0274, an ester prodrug of a metabotropic glutamate receptor 2/3 agonist with improved oral bioavailability. Eur J Med Chem. 203:112521.
- Watanabe M, Marcy B, Kinoshita K, Fukasawa M, Hikichi H, Chaki S, Okuyama S, Gevorkyan H, Yoshida S. 2020. Safety and pharmacokinetic profiles of MGS0274 besylate (TS-134), a novel metabotropic glutamate 2/3 receptor agonist prodrug, in healthy subjects. Br J Clin Pharmacol. 86(11):2286–2301.
- Williams ET, Bacon JA, Bender DM, Lowinger JJ, Guo W-K, Ehsani ME, Wang X, Wang H, Qian Y-W, Ruterbories KJ, et al. 2011. Characterization of the expression and activity of carboxylesterases 1 and 2 from the beagle dog, cynomolgus monkey, and human. Drug Metab Dispos. 39(12):2305–2313.
- Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. 2007. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 8(7):676–684.