
Abstract
Polyglutamine (polyQ) expansion within Huntingtin (Htt) causes the fatal neurodegenerative disorder Huntington’s Disease (HD). Although Htt is ubiquitously expressed and conserved from Drosophila to humans, its normal biological function is still being elucidated. Here we characterize a role for the Drosophila Htt homolog (dHtt) in fast axonal transport (FAT). Generation and expression of transgenic dHtt-mRFP and human Htt-mRFP fusion proteins in Drosophila revealed co-localization with mitochondria and synaptic vesicles undergoing FAT. However, Htt was not ubiquitously associated with the transport machinery, as it was excluded from dense-core vesicles and APLIP1 containing vesicles. Quantification of cargo movement in dHtt deficient axons revealed that mitochondria and synaptic vesicles show a decrease in the distance and duration of transport, and an increase in the number of pauses. In addition, the ratio of retrograde to anterograde flux was increased in mutant animals. Dense-core vesicles did not display similar defects in processivity, but did show altered retrograde to anterograde flux along axons. Given the co-localization with mitochondria and synaptic vesicles, but not dense-core vesicles, the data suggest dHtt likely acts locally at cargo interaction sites to regulate processivity. An increase in dynein heavy chain expression was also observed in dHtt mutants, suggesting that the altered flux observed for all cargo may represent secondary transport changes occurring independent of dHtt’s primary function. Expression of dHtt in a milton (HAP1) mutant background revealed that the protein does not require mitochondria or HAP1 to localize along axons, suggesting Htt has an independent mechanism for coupling with motors to regulate their processivity during axonal transport.
Introduction
How polyglutamine (polyQ) expansion within the Huntingtin (Htt) protein causes Huntington’s disease (HD) is poorly understood. HD is a member of a family of late-onset neurodegenerative disorders associated with polyQ expansion. Although protein aggregation of the polyQ protein is commonly observed, the causative mechanism of pathogenesis is unclear. The polyQ disorders present with distinct neurological phenotypes, though the affected proteins are expressed throughout the brain and other non-neuronal tissues. These observations suggest that polyQ expansion may not only cause a dominant gain-of-function phenotype, but also alter properties of the associated protein that disrupt its function in specific neuronal populations (Kratter & Finkbeiner, Citation2010; Orr, Citation2012). Increasing evidence has implicated Htt in intracellular transport and specifically in fast axonal transport (FAT) (Li & Conforti, Citation2013; Reddy & Shirendeb, Citation2012). One model of HD pathology suggests that pathogenic Htt disrupts FAT, leading to cumulative insults that confer neuronal specificity and late-onset symptoms. Several other adult-onset neurodegenerative disorders have been associated with defects in axonal transport (Perlson, Maday, Fu, Moughamian, & Holzbaur, Citation2010), suggesting FAT defects may be a common contributor to neurodegenerative pathology.
Biochemical studies are consistent with Htt functioning in intracellular transport. Htt binds HAP1 (Li et al., Citation1995), a known regulator of FAT via its interaction with kinesin (McGuire, Rong, Li, & Li, Citation2006) and dynein (Engelender et al., Citation1997). Additionally, the N-terminal 17 amino acids of Htt confer membrane binding and interactions with mitochondria, Golgi, and other membrane-bound organelles (Rockabrand et al., Citation2007). A direct interaction between Htt and the dynein/dynactin complex has also been described (Caviston, Zajac, Tokito, & Holzbaur, Citation2011). Knockout of htt in mice leads to early embryonic lethality (Nasir et al., Citation1995). However, lethality is rescued by wild-type extraembyronic expression, and a role for Htt in transport of ferric ions to the embryo has been suggested (Dragatsis, Efstratiadis, & Zeitlin, Citation1998). Experiments in murine striatal neuron culture suggest that knockdown of Htt alters mitochondrial transport, with an increase in pauses and a decrease in processivity and velocity (Trushina et al., Citation2004). Htt has also been implicated in transport of the neurotrophic factor BDNF and its receptor TrkB (Gauthier et al., Citation2004; Liot et al., Citation2013; Zuccato & Cattaneo, Citation2007), as well as autophagosomes (Wong & Holzbaur, Citation2014). Our results extend these findings by examining null mutants in the Drosophila Htt homolog (dHtt) to test Htt function in vivo. We find that dHtt co-localizes with a subset of axonal cargo and that dhtt−/− mutants display defects in FAT that can be rescued with Drosophila and nonpathogenic human Htt transgenes, but not with a pathogenic human polyQ Htt transgene. Both Drosophila and human nonpathogenic Htt, but not pathogenic Htt, can rescue the transport phenotypes, suggesting an ancient and conserved role for Htt as a regulator of axonal transport.
Materials and methods
Generation of Htt constructs
Transgenic animals expressing HttQ15-mRFP and HttQ138-mRFP were previously described (Weiss, Kimura, Lee, & Littleton, Citation2012). A cDNA encoding the N-terminal 2 kb of dhtt was cloned using the Gateway cloning system (Invitrogen, Carlsbad, CA). The N-terminal 2 kb of dhtt was PCR-amplified from a cDNA library, subcloned into the pENTR-dTOPO vector, and recombined into the pTWR destination vector with a C-terminal mRFP tag and the UAS promoter.
Drosophila stocks and genetics
The dhtt null line was generated as previously reported (Zhang, Feany, Saraswati, Littleton, & Perrimon, Citation2009) by inserting a CG9990 genomic rescue construct into the Df(93E2) background, which lacks both dhtt and CG9990. The C380-GAL4 driver was used in this study due to its high expression level in all motor axons. The CCAP-GAL4 driver line was used to image SV and DCV transport (courtesy of John Ewer). Drosophila lines C380-GAL4, cytochrome c oxidase-GFP (Mito-GFP), atrial natriuretic factor-GFP (ANF-GFP), and synaptotagmin-1-GFP (Syt-1-GFP) were obtained from the Bloomington Fly Stock Center, (Bloomington, IN). All crosses were maintained at 25 °C on standard medium.
Live imaging and transport analysis
Organelle runs, pauses, and velocities were measured in freshly dissected third instar larvae pinned to a Sylgard covered slide immersed in HL3 insect ringer with 0.2 mM Ca2+ using a Zeiss upright scanning confocal microscope. Flux and long-term transport analyses were measured in third instar larvae anesthetized with Suprane in a custom chamber that allowed imaging of fluorescently tagged constructs through the cuticle for up to 3 h (Fuger, Behrends, Mertel, Sigrist, & Rasse, Citation2007). Both transport protocols employed imaging at one frame per second (fps) for mitochondria, and three fps for SVs and DCVs, for a total of six minutes, across a bleached 50 μm region of motor neuron axon using a 60× objective. The CCAP-GAL4 driver, which expresses in a single neuron per hemisegment, was utilized to track single vesicles. Imaging was done in the distal 200 μm of axons innervating muscles 6/7 or 12/13 in segments A3 or A4. Individual organelles moving into the bleached region of an axon were marked with a cursor in the ImageJ plugin MtrackJ (NIH, Bethesda, MD) to indicate x, y, t coordinates and movement parameters were calculated as previously described (Louie, Russo, Salkoff, Wellington, & Zinsmaier, Citation2008). For flux analysis, individual organelles were tracked with the Volocity Imaging software (Perkin Elmer, Waltham, MA). Manual tracking was done for a minimum of 60 s per organelle to calculate all other movement parameters. An one-way ANOVA followed by Tukey’s post-hoc test were used for group analysis and Student’s t test for individual statistics. Statistical values were calculated using Microsoft Excel and Origin Graphing Software (Northampton, MA).
Western blot analysis and immunocytochemistry
Monoclonal anti-kinesin heavy chain (SUK4) or anti-dynein heavy chain (2C-11) (Developmental Studies Hybridoma Bank, Iowa City, IA) antibodies were used at 1:100 to probe adult head extracts. Blots were visualized on a Li-Cor Odyssey Infrared Imaging System (Lincoln, NE) and quantified with the accompanying software. For HRP staining, third instar larvae expressing UAS-mito-GFP or UAS-Syt-1-GFP with the C380-GAL4 driver were reared at 25 °C and dissected in HL3 insect ringer. Images were captured with a Zeiss Pascal laser scanning confocal microscope (Carl Zeiss MicroImaging Inc., Jena, Germany) using the accompanying Zeiss PASCAL software. Area and number of puncta were measured using ImageJ channel area measurement functions and the ICTN counter plugin.
Results
Drosophila and human Htt N-terminal fragments co-localize with a subset of axonal cargo
To characterize the subcellular localization and properties of dHtt, we generated transgenic animals expressing a UAS-mRFP-tagged N-terminal fragment of dHtt for in vivo imaging in third instar Drosophila larvae. We used the N-terminal region of dHtt containing 667 amino acids that encompasses a region of high homology between human and dHtt (Li, Karlovich, Fish, Scott, & Myers, Citation1999). This fragment is similar in size to the human Htt N-terminal caspase-6 cleavage fragment that has been well characterized in Drosophila (Weiss et al., Citation2012). We co-expressed dHtt-mRFP or human Htt with a nonpathogenic 15 polyQ repeat tagged with mRFP (HttQ15-mRFP) with GFP-tagged transport cargos in Drosophila motor neurons using the C380-GAL4 driver. Both Drosophila and human Htt proteins were abundant in motor axons and underwent FAT in both anterograde and retrograde directions. dHtt and HttQ15 showed robust co-localization with mitochondria, moderate co-localization with synaptic vesicles (SVs) (∼50%) and little to no co-localization with dense core vesicles (DCV) or cargos marked by the JIP-1-like protein APLIP-1 ()). Because dHtt and human Htt co-localize with only a subset of axonal cargos, it is unlikely that Htt functions as a ubiquitous component of the axonal transport machinery.
Figure 1. Localization of Drosophila and human Htt N-terminal fragments with transport cargo. (A) GFP-tagged cargos were expressed with C380-GAL4 with either an N-terminal 665 amino acid fragment of dHtt (left panel), or an N-terminal 588 amino acid fragment of normal (Q15, middle panel) or pathogenic (Q138, right panel) human Htt. Each of the 12 image squares depicts three image channels of the same axon: top channel = Htt-RFP; middle channel = GFP-cargo, bottom = merged. Scale bar =10 μm. (B) Calculation of RFP and GFP fluorescence intensity overlap using Pearson’s coefficient (R value) calculated with Volocity software. *denotes p < 0.05.
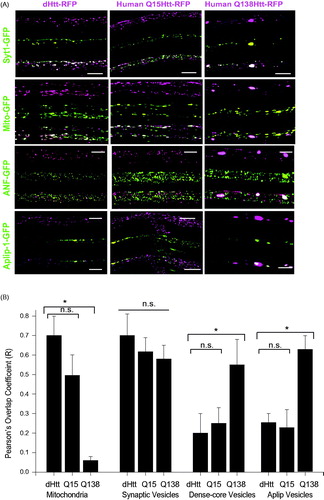
We next characterized the co-localization of axonal cargos with human Htt expressing a pathogenic 138 polyQ repeat tagged with mRFP (HttQ138-RFP) (). HttQ138 formed large aggregates within axons that were immobile over 3 h imaging sessions (Weiss et al., Citation2012), similar to our prior observations with untagged pathogenic Htt proteins (Lee, Yoshihara, & Littleton, Citation2004). In addition to aggregates, smaller HttQ138 particles were observed that underwent FAT similar to dHtt and HttQ15. These HttQ138 transport particles showed a strong reduction in co-localization with mitochondria compared to HttQ15 or dHtt (), quantified using the Pearson’s correlation coefficient for overlap (). Co-localization of HttQ138 transport packets with SVs was not altered. In contrast to motile HttQ138 transport particles, there was increased co-localization between immobile HttQ138 aggregates and DCVs, SVs, and cargos labeled by APLIP-1-GFP ()), consistent with HttQ138 aggregates trapping axonal cargo.
Drosophila lacking dhtt display defects in FAT
Drosophila has been well established as a model for analysis of mitochondrial FAT (Hollenbeck & Saxton, Citation2005). We used null mutants in the dhtt locus (Zhang et al., Citation2009) to determine if Htt regulates axonal transport. RNAi-mediated knockdown of dhtt was reported to disrupt transport and cause axonal traffic jams consisting of concentrated accumulations of SVs (Gunawardena et al., Citation2003). However, staining for SV protein distribution in the dhtt null (dhtt−/−) mutant did not reveal cargo accumulation along axons, suggesting previous observations were likely secondary to off-target effects by RNAi hairpins. We observed that all cargos analyzed in dhtt−/−, including mitochondria, SVs, endosomes, and DCVs, moved bi-directionally within axons, with no accumulation during live imaging sessions.
To determine if FAT defects beyond cargo accumulation are associated with loss of dHtt, mitochondria were tracked manually with ImageJ (NIH, Bethesda, MD) and the object tracking function in the Volocity 4-D imaging software package (Perkin Elmer, Waltham, MA). We observed no change in the number of motile mitochondria entering axonal regions following FRAP in dhtt−/− and controls. However, kymographs of mitochondrial movement over a 50 μm region of axon for 300 s revealed an altered ratio of retrograde to anterograde moving mitochondria in dhtt−/− (). While control larvae have a 1:1 ratio of retrograde to anterograde mitochondrial flux, dhtt−/− animals show an increased 1.85:1 retrograde to anterograde ratio (p < 0.05) (N ≥ 10 larvae, n ≥ 10 mitochondria per larvae for all measurements). This alteration in mitochondrial transport results from a decrease in anterograde flux (control = 10 ± 1.8 min−1; dhtt−/− = 7.5 ± 1.1 min−1) and an increase in retrograde flux (control = 9.8 ± 1.1 min−1; dhtt−/− = 13.9 ± 1.4 min−1).
Figure 2. Abnormal flux of mitochondria in dhtt−/− mutants. (A) Kymographs of mitochondrial movement in motor axons. (B) Quantification of retrograde: anterograde flux. Drosophila full-length (FL) Htt and an N-terminal mRFP tagged Drosophila dHtt construct rescued the increased flux in dhtt−/−. Human (Hu) Htt Q16 also rescued the transport defect, while human HttQ138 did not. All rescue constructs were assayed in the dhtt−/− null background. The Drosophila full-length (FL) Htt construct was driven with the endogenous dhtt promoter and all others were driven with C380-GAL4. Error bars represent SEM, *indicates p < 0.05 ANOVA. n.s.: no significant difference.
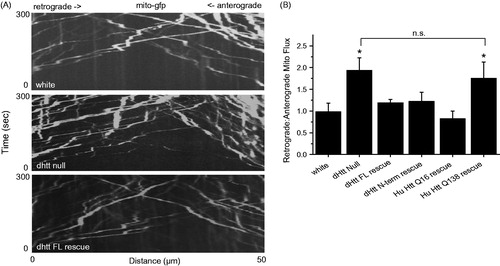
The altered flux of mitochondria in dhtt−/− was used to assay the ability of transgenically expressed Htt proteins to rescue FAT defects in the dhtt−/− background. The flux defect was rescued by expression of a minigene containing full-length dhtt under the control of its endogenous regulatory region (flux ratio of 1.22:1; p < 0.05 compared to dhtt−/−, p > 0.5 compared to control) (), indicating FAT defects arise secondary to loss of dHtt. The transport defects were also rescued by expression of a full-length human HttQ16 transgene (Romero et al., Citation2008) driven by C380-GAL4 (flux ratio of 0.82:1; p < 0.05 compared to dhtt−/−; p > 0.5 compared to control) (). Expression of the N-terminal 667 amino acid dHtt-mRFP transgenic protein used to characterize cargo co-localization also rescued the flux phenotype (flux ratio of 1.25:1; p < 0.05 compared to dhtt−/−, p > 0.5 compared to control), suggesting the activity required to regulate mitochondrial transport resides in the highly conserved N-terminal region of Htt. In contrast to dHtt or human nonpathogenic Htt, the flux phenotype was not rescued by a human HttQ138 transgene (flux ratio of 1.76:1; p > 0.5 compared to dhtt−/−, p < 0.05 compared to control) (), suggesting a loss-of-function associated with the expanded polyQ tract in the Htt protein. These results are consistent with the observation that HttQ138 transport particles show reduced co-localization with mitochondria ().
dHtt regulates mitochondrial processivity
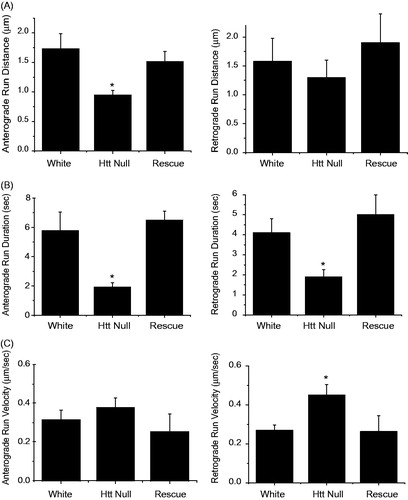
To further examine the effects of dHtt on axonal transport, individual mitochondria were tracked manually during time-lapse imaging to determine run distance, run duration, and velocity (). Similar manual tracking of mitochondria has revealed transport defects in several Drosophila mutants (Pilling, Horiuchi, Lively, & Saxton, Citation2006). Loss of dHtt altered several FAT parameters, including a decrease in run distance and duration for both anterograde and retrograde mitochondrial movements ()). These alterations in FAT indicate motor output or cargo processivity is decreased in the absence of dHtt. The FAT phenotypes were rescued by expression of a minigene containing full-length dhtt (p < 0.05 compared to dhtt−/−; p > 0.5 compared to control) ()). Loss of dHtt also led to increased run velocity for retrograde moving mitochondria, which represents an average of instantaneous velocity measurements while a mitochondrion is in motion ().
Figure 3. FAT defects for mitochondria in dhtt−/− mutants. Run data was collected from individual mitochondria tracked in each one-second frame of a 300 image confocal time series. Tracks were used to calculate run distance, duration, and velocity for control white larvae, dhtt−/−, and dhtt−/− expressing a dhtt full-length rescue construct. Anterograde or retrograde designations reflect the imprinted or primary direction of movement. (A) dhtt−/− larvae exhibit a decrease in run distance for both anterograde and retrograde moving mitochondria. (B) dhtt−/− larvae exhibit a decrease in run duration for both anterograde and retrograde moving mitochondria. (C) dhtt−/− larvae exhibit an increase in velocity for retrograde moving mitochondria. Error bars represent SEM, *indicates p < 0.05.
Differential effects of dHtt on FAT of synaptic vesicles and dense-core vesicles
To determine if dHtt’s effects on FAT were specific to mitochondria, we analyzed the kinetics of transport for synaptic and DCVs in dhtt−/− larvae. SVs co-localize with dHtt along axons, and we found that SV transport kinetics were disrupted in dhtt−/−, displaying similar alterations as observed for mitochondrial transport (). However, DCVs, which did not co-localize with dHtt, displayed normal local transport kinetics, including run distance, duration, and velocity (). In contrast to these FAT parameters, the increased retrograde to anterograde flux observed for mitochondrial transport was also observed for DCVs (). These findings indicate dHtt acts locally to regulate transport kinetics of co-localized organelles, while the alteration in retrograde to anterograde flux is secondary to a more global defect in transport.
Table 1. Summary of transport kinetics for live tracking of synaptic vesicles (Syt-1-GFP) and dense-core vesicles (ANF-GFP) expressed with the CCAP-GAL4 driver.
Track analysis of mitochondrial kinetics reveal defects in cargo attachment and/or motor coordination
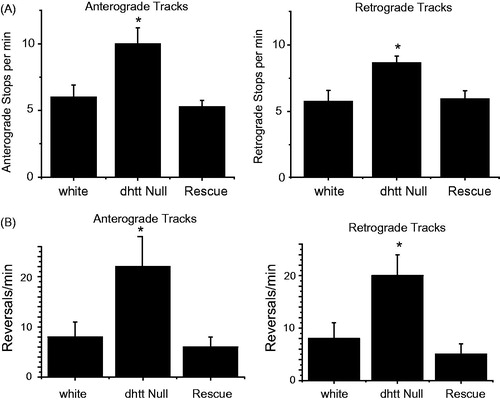
Our previous measurements of FAT kinetics measured mitochondria in motion. In contrast, track analysis examines the entire path of an individual mitochondrion from the time it enters a bleached region of axon until it leaves on the opposite side. As such, track analysis provides information on all runs, stops, and reversals, generating a measure for motor processivity and coordination over a 50 μm range. Mitochondria are imprinted with a specific directionality over this distance, either primarily retrograde or anterograde, while occasionally pausing or briefly reversing. Measuring track movements, we observed an increase in the frequency of stops for anterograde tracks in dhtt−/− (10.0 ± 1.2 min−1) compared to control (6.0 ± 0.9 min−1), and an increase in the frequency of stops for retrograde tracks in dhtt−/− larvae (8.3 ± 0.5 min−1) compared to control (5.5 ± 0.8 min−1) (p < 0.05, N ≥ 10 larvae, n ≥ 10 mitochondria per larvae), indicating a defect in processivity (). This stopping defect was rescued by expression of a dhtt rescue minigene construct. Similarly, we also measured an increase in the frequency of reversals for anterograde moving mitochondria in dhtt−/− (22 ± 5.9 min−1) compared to controls (8 ± 3 min−1; p < 0.05) and for retrograde moving mitochondria in dhtt−/− (20 ± 4 min−1) compared to controls (8 ± 3 min−1; p < 0.05) (), indicating a defect in cargo coordination or attachment. Expression of the dhtt minigene also rescued the defects in reversal. We observed similar defects for SV axonal transport, but not for DCVs (), again indicating dHtt acts locally to regulate co-localized cargo transport.
Figure 4. Track analysis for anterograde and retrograde moving mitochondria in control white larvae, dhtt−/−, and dhtt−/− expressing a dhtt full-length rescue construct. Tracks represent three states of mitochondrial movement for an individual mitochondrion tracked over the entire track length, including movement in the primary direction, reverse direction, and stops. Anterograde or retrograde designations reflect the imprinted or primary direction of movement. (A) Loss of dHtt results in an increase in mitochondrial stops, indicating a defect in processivity. (B) Loss of dHtt results in an increase in mitochondrial run reversals, indicating a defect in motor coordination. N > 10 larvae per genotype and n > 12 mitochondria per larvae for all panels. Error bars represent SEM, *indicates p < 0.05.
HAP1 and mitochondria are not required for dHtt localization
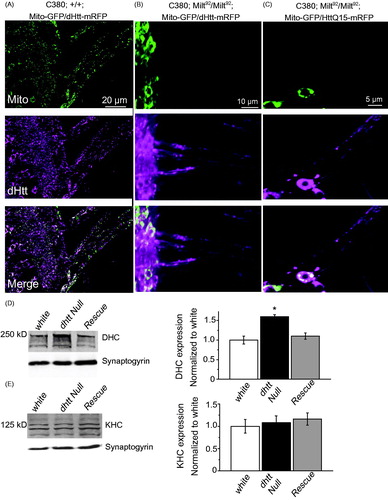
To examine interactions required for the formation of the Htt-motor-cargo complex, we tested if the interaction between dHtt and its binding partner HAP1 was necessary for dHtt transport along axons. The Drosophila ortholog of HAP1, Milton, is required for transport of mitochondria into axons (Stowers, Megeath, Górska-Andrzejak, Meinertzhagen, & Schwarz, Citation2002) (). In milton LOF mutants (milt92), mitochondria are concentrated in the cell body and do not enter axons. Despite the lack of HAP1 or mitochondria in the axon, both dHtt and human HttQ15 localized to axons ()). Both human and Drosophila N-terminal fragments are competent to bind HAP1, as the HAP1 binding domain of Htt resides within the first 230 amino acids of Htt (Li et al., Citation1995). These data demonstrate that dHtt undergoes axonal transport independent of the presence of HAP1 or mitochondria in the neuronal process. This suggests that other adapter proteins or direct interactions are necessary to mediate Htt-motor interactions.
Figure 5. Mitochondria and HAP1 are not required for Htt axonal localization. (A) Expression of Mito-GFP and dHtt-mRFP in motor neurons in a control stage 17 embryonic ventral nerve cord shows distribution of mitochondria and Htt throughout the cell body and from axons exiting the CNS. (B) Without HAP1 in a milt92 mutant background, mitochondria are trapped within the cell body, while dHtt-mRFP is still present in axons. (C) Human HttQ15-mRFP also localizes to axons in the absence of HAP1. (D) Dynein heavy chain levels are increased in dhtt−/− adults. Adult head extracts from ten 20-day old flies were assayed by Western blot and probed for dynein heavy chain (DHC) and Synaptogyrin (loading control). Quantification of DHC levels from five separate measurements normalized to control white animals is shown in the right panel. (E) The levels of kinesin heavy chain (KHC) were not changed in dhtt−/− aged adults. Quantification of KHC levels from five separate measurements normalized to control white animals is shown on the right.
Dense-core vesicles display defective flux along axons
To determine if loss of Htt alters additional aspects of axonal transport, we assayed for changes in transport kinetics of ANF-tagged DCVs, which do not co-localize with dHtt (, ). Velocity, run length, stoppage, and reversals for DCVs were not altered in dhtt−/−, suggesting dHtt regulates cargo processivity locally, likely mediated by direct interactions between dHtt, cargos, motors, and/or other adapter proteins. However, DCVs displayed a similar flux phenotype as observed for mitochondria and SVs, with an increase in the ratio of retrograde to anterograde moving cargo (). To determine if loss of dHtt altered the levels of dynein and kinesin motors that could underlie more global transport phenotypes, we assayed their levels by western blot analysis. We observed that the levels of dynein heavy chain are increased by 50% in 20-day old dhtt−/− adults compared to control or rescued animals (). In contrast, kinesin heavy chain levels were unaltered (). Increased dynein levels have also been observed in the striatum of 12 month-old mice heterozygous for Q175-Htt (Smith et al., Citation2014), indicating the loss of Htt and polyQ expansion within the protein may impact overall retrograde transport properties. These data indicate loss of dHtt results in increased dynein levels, which may contribute to the increase in retrograde flux of FAT cargos, independent of the specific effects of local Htt on cargo processivity.
Discussion
It is largely unclear how Htt normally regulates neuronal function, and whether disruptions in this process contribute to HD (Schulte & Littleton, Citation2011). Previous studies have implicated Htt in a variety of biological processes, including neurogenesis (Ben M’Barek et al., Citation2013), mitotic spindle orientation (Godin et al., Citation2010), chromatin organization (Dietz et al., Citation2015; Seong et al., Citation2010), macroautophagy (Rui et al., Citation2015), iron import (Lumsden, Henshall, Dayan, Lardelli, & Richards, Citation2007) cargo transport (Gauthier et al., Citation2004; Trushina et al., Citation2004) and synaptic plasticity (Choi et al., Citation2014). Here we demonstrate that Drosophila dhtt−/− mutants display defects in axonal transport of cargos in vivo. The data suggest dHtt functions as a positive regulator of FAT for mitochondria and SVs, as loss of dHtt results in increased stops and decreased run lengths for these organelles. dHtt regulates cargo processivity locally, as these defects are only observed for organelles that co-localize with dHtt. Given dHtt simultaneously exerts a negative regulatory role in velocity and a positive role in processivity, we hypothesize that dHtt may act as a scaffold to coordinate the activity of other positive and negative regulators of organelle movement, consistent with observations from previous studies (Fu & Holzbaur, Citation2014).
Is this normal role of Htt in axonal transport disrupted by pathogenic polyQ expansion within the protein Htt? The reduced co-localization between human HttQ138 and mitochondria that we observe is consistent with a loss-of-function transport defect due to polyQ expansion. The observation that transport of BDNF in mammalian systems is disrupted by polyQ expansion within Htt (Gauthier et al., Citation2004; Pardo et al., Citation2010) is consistent with our observations on mitochondrial transport, and suggests pathogenic Htt may alter FAT properties. Recent studies have also implicated Htt in the transport of several cargo in both axons and dendrites, indicating Htt may play a widespread role in regulating transport within neurons (Liot et al., Citation2013; Roux et al., Citation2012; Zala, Hinckelmann, & Saudou, Citation2013). The similar co-localization patterns observed between Drosophila and human Htt with axonal cargos (), and the ability of human Htt to rescue dhtt−/− defects in mitochondrial flux (), suggest Drosophila and human Htt may share similar roles in axonal transport. Functional redundancy between Drosophila and human Htt has also been demonstrated in mitotic spindle orientation (Godin et al., Citation2010) and BDNF transport (Zala et al., Citation2013).
How does Htt control axonal transport? Significant effects on reversals suggest changes in motor coordination (). Transport kinetics in dhtt mutants is altered for both anterograde and retrograde moving organelles, suggesting that dHtt regulates both kinesin and dynein mediated transport. Bidirectional transport mechanisms are not well defined, but the ability to coordinate anterograde and retrograde movement of motors on a particular organelle is essential. Current data indicate that kinesin and dynein motors are both present on organelles such as mitochondria and SVs (Ligon, Tokito, Finklestein, Grossman, & Holzbaur, Citation2004; De Vos, Sable, Miller, & Sheetz, Citation2003), indicating that a yet undefined mechanism is required to coordinate motor movement. Htt has been shown to interact indirectly with both kinesin (McGuire et al., Citation2006) and dynein/dynactin (Engelender et al., Citation1997) through its interaction with HAP1. These in vivo findings agree with cell culture experiments demonstrating that phosphorylation of Htt serine 421 favors anterograde transport, while dephosphorylated Htt favors retrograde transport, suggesting Htt acts as a ‘molecular switch’ in controlling transport (Colin et al., Citation2008). In addition, regulators of the JNK pathway have also been implicated in coordinating kinesin and dynein activation, suggesting Htt could interface with JNK signaling to regulate processivity by acting as a scaffolding protein or regulator of the JNK pathway (Morfini et al., Citation2009).
Defects similar to what we observe in dHtt mutants have been seen following disruption of dynein regulatory proteins, which cause similar increases in pausing and reversals. For example, loss of the dynactin subunit Arp1 increases pausing and reversals for anterograde and retrograde reversals (Haghnia et al., Citation2007). In contrast, reduced expression of the dynactin subunit glued increases velocity for anterograde and retrograde mitochondria (Pilling et al., Citation2006), similar to what we observe in dhtt−/−. The loss of adapter proteins that link cargos to motors have also been found to alter FAT kinetics. Loss of the mitochondrial adapter protein, Miro, causes increased pausing, decreased run distance, and decreased velocity for anterograde and retrograde moving mitochondria (Guo et al., Citation2005). Mutation of the blue cheese locus, which encodes a putative lysosomal transport adapter, results in an increased retrograde to anterograde flux, and increased reversals for lysosomes, but not mitochondria (Lim & Kraut, Citation2009). These animals also exhibit decreased lifespan and brain degeneration (Finley et al., Citation2003), as seen in dHtt mutants (Zhang et al., Citation2009). Thus, transport defects observed in dHtt mutants overlap phenotypes observed in conditions of decreased motor activity, reduced motor regulatory function and disrupted cargo attachment.
In conclusion, our results suggest Htt interfaces with transport motors and membrane-bound organelles to coordinate bi-directional axonal transport. The observation that nonpathogenic human Htt can rescue axonal transport defects in dhtt mutants, while pathogenic Htt cannot, hints that this property may be conserved beyond Drosophila and may contribute to defects observed in HD patients. As future treatments for HD may ultimately involve direct manipulation of Htt activity, assays that provide readout of the protein’s biological activity within neurons, like the Drosophila FAT phenotypic assays, will be useful for testing potential therapeutics. Any inhibitor that prevents Htt polyQ pathogenesis, but that disrupts normal Htt function, would have to be evaluated more carefully. In contrast, reagents that can block the abnormal function of pathogenic polyQ-containing Htt proteins, without compromising the required role of endogenous Htt in axonal transport would be especially promising for further development.
Funding information
This work was supported by NIH grant NS40296 to J. Troy Littleton and NIH Pre-Doctoral Training Grant T32GM007287 to Kurt R. Weiss.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Caviston, J.P., Zajac, A.L., Tokito, M., & Holzbaur, E.L. (2011). Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Molecular Biology of the Cell, 22, 478–492.
- Choi, Y.B., Kadakkuzha, B.M., Liu, X.A., Akhmedov, K., Kandel, E.R., & Puthanveettil, S.V. (2014). Huntingtin is critical both pre- and postsynaptically for long-term learning-related synaptic plasticity in aplysia. PLoS One, 9, e103004.
- Colin, E., Zala, D., Liot, G., Rangone, H., Borrell-Pagès, M., Li, X.J. … Saudou, F. (2008). Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. The EMBO Journal, 27, 2124–2134.
- Dietz, K.N., Di Stefano, L., Maher, R.C., Zhu, H., Macdonald, M.E., Gusella, J.F., & Walker, J.A. (2015). The Drosophila Huntington’s disease gene ortholog dhtt influences chromatin regulation during development. Human Molecular Genetics, 24, 330–345.
- Dragatsis, I., Efstratiadis, A., & Zeitlin, S. (1998). Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development, 125, 1529–1539.
- Engelender, S., Sharp, A.H., Colomer, V., Tokito, M.K., Lanahan, A., Worley, P. … Holzbaur, E. L. (1997). Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Human Molecular Genetics, 6, 2205–2212.
- Finley, K.D., Edeen, P.T., Cumming, R.C., Mardahl-Dumesnil, M.D., Taylor, B.J., Rodriguez, M.H. … Hwang, C.E. (2003). blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. The Journal of Neuroscience, 23, 1254–1264.
- Fu, M.M., & Holzbaur, E.L. (2014). Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends in Cell Biology, 24, 564–574.
- Fuger, P., Behrends, L.B., Mertel, S., Sigrist, S.J., & Rasse, T.M. (2007). Live imaging of synapse development and measuring protein dynamics using two-color fluorescence recovery after photo-bleaching at Drosophila synapses. Nature Protocols, 2, 3285–3298.
- Gauthier, L.R., Charrin, B.C., Borrell-Pagès, M., Dompierre, J. P., Rangone, H., Cordelières, F.P. … De Mey, J. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell, 118, 127–138.
- Godin, J.D., Colombo, K., Molina-Calavita, M., Keryer, G., Zala, D., Charrin, B.C. … Dietrich, P. (2010). Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron, 67, 392–406.
- Gunawardena, S., Her, L.S., Brusch, R.G., Laymon, R.A., Niesman, I.R., Gordesky-Gold, B. … Sintasath, L. (2003). Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron, 40, 25–40.
- Guo, X., Macleod, G.T., Wellington, A., Hu, F., Panchumarthi, S., Schoenfield, M. … Marin, L. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron, 47, 379–393.
- Haghnia, M., Cavalli, V., Shah, S.B., Schimmelpfeng, K., Brusch, R., Yang, G. … Herrera, C. (2007). Dynactin is required for coordinated bidirectional motility, but not for dynein membrane attachment. Molecular Biology of the Cell, 18, 2081–2089.
- Hollenbeck, P.J., & Saxton, W.M. (2005). The axonal transport of mitochondria. Journal of Cell Science, 118, 5411–5419.
- Kratter, I.H., & Finkbeiner, S. (2010). PolyQ disease: Too many Qs, too much function? Neuron, 67, 897–899.
- Lee, W.C., Yoshihara, M., & Littleton, J.T. (2004). Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proceedings of the National Academy of Sciences of the United States of America, 101, 3224–3229.
- Li, J.Y., & Conforti, L. (2013). Axonopathy in Huntington’s disease. Experimental Neurology, 246, 62–71.
- Li, X.J., Li, S.H., Sharp, A.H., Nucifora, F.C., Schilling, G., Lanahan, A. … Worley, P. (1995). A huntingtin-associated protein enriched in brain with implications for pathology. Nature, 378, 398–402.
- Li, Z., Karlovich, C.A., Fish, M.P., Scott, M.P., & Myers, R.M. (1999). A putative Drosophila homolog of the Huntington’s disease gene. Human Molecular Genetics, 8, 1807–1815.
- Ligon, L.A., Tokito, M., Finklestein, J.M., Grossman, F.E., & Holzbaur, E.L. (2004). A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. The Journal of Biological Chemistry, 279, 19201–19208.
- Lim, A., & Kraut, R. (2009). The Drosophila BEACH family protein, blue cheese, links lysosomal axon transport with motor neuron degeneration. The Journal of Neuroscience, 29, 951–963.
- Liot, G., Zala, D., Pla, P., Mottet, G., Piel, M., & Saudou, F. (2013). Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. The Journal of Neuroscience, 33, 6298–6309.
- Louie, K., Russo, G.J., Salkoff, D.B., Wellington, A., & Zinsmaier, K.E. (2008). Effects of imaging conditions on mitochondrial transport and length in larval motor axons of Drosophila. Comparative Biochemistry and Physiology. Part A, Molecular & Integrative Physiology, 151, 159–172.
- Lumsden, A.L., Henshall, T.L., Dayan, S., Lardelli, M.T., & Richards, R.I. (2007). Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Human Molecular Genetics, 16, 1905–1920.
- Ben M’Barek, K., Pla, P., Orvoen, S., Benstaali, C., Godin, J.D., Gardier, A.M. … Saudou, F. (2013). Huntingtin mediates anxiety/depression-related behaviors and hippocampal neurogenesis. The Journal of Neuroscience, 33, 8608–8620.
- McGuire, J.R., Rong, J., Li, S.H., & Li, X.J. (2006). Interaction of Huntingtin-associated protein-1 with kinesin light chain: Implications in intracellular trafficking in neurons. The Journal of Biological Chemistry, 281, 3552–3559.
- Morfini, G.A., You, Y.M., Pollema, S.L., Kaminska, A., Liu, K., Yoshioka, K. … Brady, S.T. (2009). Pathogenic huntingtin inhibits faxt axonal transport by activating JNK3 and phosphorylating kinesin. Nature Neuroscience, 12, 864–871.
- Nasir, J., Floresco, S.B., O’Kusky, J.R., Diewert, V.M., Richman, J.M., Zeisler, J. … Borowski, A. (1995). Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell, 81, 811–823.
- Orr, H.T. (2012). Polyglutamine neurodegeneration: Expanded glutamines enhance native functions. Current Opinion in Genetics & Development, 22, 251–255.
- Pardo, R., Molina-Calavita, M., Poizat, G., Keryer, G., Humbert, S., & Saudou, F. (2010). pARIS-htt: An optimised expression platform to study huntingtin reveals functional domains required for vesicular trafficking. Molecular Brain, 3, 17.
- Perlson, E., Maday, S., Fu, M.M., Moughamian, A.J., & Holzbaur, E.L. (2010). Retrograde axonal transport: Pathways to cell death? Trends in Neurosciences, 33, 335–344.
- Pilling, A.D., Horiuchi, D., Lively, C.M., & Saxton, W.M. (2006). Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Molecular Biology of the Cell, 17, 2057–2068.
- Reddy, P.H., & Shirendeb, U.P. (2012). Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochimica et Biophysica Acta, 1822, 101–110.
- Rockabrand, E., Slepko, N., Pantalone, A., Nukala, V.N., Kazantsev, A., Marsh, J.L. … Sullivan, P.G. (2007). The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Human Molecular Genetics, 16, 61–77.
- Romero, E., Cha, G.H., Verstreken, P., Ly, C.V., Hughes, R.E., Bellen, H.J., & Botas, J. (2008). Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron, 57, 27–40.
- Roux, J.C., Zala, D., Panayotis, N., Borges-Correia, A., Saudou, F., & Villard, L. (2012). Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway. Neurobiology of Disease, 45, 786–795.
- Rui, Y.N., Xu, Z., Patel, B., Chen, Z., Chen, D., Tito, A. … David, G. (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nature Cell Biology, 17, 262–275.
- Schulte, J., & Littleton, J.T. (2011). The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Current Trends in Neurology, 5, 65–78.
- Seong, I.S., Woda, J.M., Song, J.J., Lloret, A., Abeyrathne, P.D., Woo, C.J. … Gregory, G. (2010). Huntingtin facilitates polycomb repressive complex 2. Human Molecular Genetics, 19, 573–583.
- Smith, G.A., Rocha, E.M., McLean, J.R., Hayes, M.A., Izen, S.C., Isacson, O., & Hallett, P.J. (2014). Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Human Molecular Genetics, 23, 4510–4527.
- Stowers, R.S., Megeath, L.J., Górska-Andrzejak, J., Meinertzhagen, I.A., & Schwarz, T.L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron, 36, 1063–1077.
- Trushina, E., Dyer, R.B., Badger, J.D., Ure, D., Eide, L., Tran, D.D. … Vrieze, B. T. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Molecular and Cellular Biology, 24, 8195–8209.
- De Vos, K.J., Sable, J., Miller, K.E., & Sheetz, M.P. (2003). Expression of phosphatidylinositol (4,5) bisphosphate-specific pleckstrin homology domains alters direction but not the level of axonal transport of mitochondria. Molecular Biology of the Cell, 14, 3636–3649.
- Weiss, K.R., Kimura, Y., Lee, W.C., & Littleton, J.T. (2012). Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington’s disease model. Genetics, 190, 581–600.
- Wong, Y.C., & Holzbaur, E.L. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. The Journal of Neuroscience, 34, 1293–1305.
- Zala, D., Hinckelmann, M.V., & Saudou, F. (2013). Huntingtin’s function in axonal transport is conserved in Drosophila melanogaster. PLoS One, 8, e60162.
- Zhang, S., Feany, M.B., Saraswati, S., Littleton, J.T., & Perrimon, N. (2009). Inactivation of Drosophila Huntingtin affects long-term adult functioning and the pathogenesis of a Huntington’s disease model. Disease Models & Mechanisms, 2, 247–266.
- Zuccato, C., & Cattaneo, E. (2007). Role of brain-derived neurotrophic factor in Huntington’s disease. Progress in Neurobiology, 81, 294–330.