
Abstract
Poly(ADP-ribose)polymerase1 (PARP1) is an important enzyme in regulating DNA replication. Inhibition of PARP1 can lead to collapsed DNA forks which subsequently causes genomic instability, making DNA more susceptible in developing fatal DNA double strand breaks. PARP1-induced DNA damage is generally repaired by homologous recombination (HR), in which BRCA2 proteins are essential. Therefore, BRCA2-deficient tumour cells are susceptible to treatment with PARP1-inhibitors (PARP1-i). Recently, BRCA2 was shown to be down-regulated by hyperthermia (HT) temporarily, and this consequently inactivated HR for several hours. In this study, we investigated whether HT exclusively interferes with HR by analysing thermal radiosensitisation of BRCA2-proficient and deficient cells. After elucidating the equitoxicity of PARP1-i on BRCA2-proficient and deficient cells, we studied the cell survival, apoptosis, DNA damage (γ-H2AX foci and comet assay) and cell cycle distribution after different treatments. PARP1-i sensitivity strongly depends on the BRCA2 status. BRCA2-proficient and deficient cells are radiosensitised by HT, indicating that HT does not exclusively act by inhibition of HR. In all cell lines, the addition of HT to radiotherapy and PARP1-i resulted in the lowest cell survival, the highest levels of DNA damage and apoptotic levels compared to duo-modality treatments. Concluding, HT not only inhibits HR, but also has the capability of radiosensitising BRCA2-deficient cells. Thus, in case of BRCA2-mutation carriers, combining HT with PARP1-i may boost the treatment efficacy. This combination therapy would be effective for all patients with PARP1-i regardless of their BRCA status.
Introduction
DNA is replicated during the S-phase of the cell cycle. Enzyme helicase unzips DNA strands, creating a replication fork, where multiple proteins can bind. These proteins can restore DNA damage thereby preventing genome instability, before DNA replication may proceed [Citation1]. If DNA replication is compromised, DNA damages, such as single strand breaks (SSBs) may occur. One of the proteins that is recruited at sites of stalled forks is Poly(ADP-ribose)polymerase1 (PARP1). Along with other repair proteins, PARP1 binds to the DNA and repairs damages and reinitiates DNA replication at the stalled forks [Citation2]. If PARP1 cannot cope with these SSBs during this semi-conservative repair process, the SSBs can be converted to DNA double strand breaks (DSBs) or also referred to as a double strand end (DSE) [Citation3]. Consequently, this primarily activates homologous recombination (HR) to repair DNA damage in the S- and G2-phases of the cell cycle [Citation4,Citation5]. Thus, as PARP1 is required for regulating replication integrity, interference by PARP1-inhibitors (PARP1-i) gives rise to DSEs caused by failures during DNA replication.
PARP1-i can induce synthetic lethality if DNA damage cannot be restored by the backup mechanism HR. For this reason, PARP1-i are under investigation for treatment of BRCA2-mutated tumours (HR-deficient tumours), as the BRCA2 proteins are essential in HR [Citation6–8].
Hyperthermia (HT), heating the tumour within a range of 40–42.5 °C for approximately 1 h, has multiple anti-cancer effects [Citation9–17], like stimulating the heat shock proteins, triggering the immune system and creating a window during which the activity of BRCA2 is suppressed by down-regulation of the protein [Citation18], resulting in an increased number of unrepaired radiation-induced DSBs [Citation19]. HT is a clinically established therapy in combination with radiotherapy or chemotherapy. Results show that HT improves local tumour control and survival [Citation20–24]. In the present study, radiosensitisation by HT was investigated in BRCA2-proficient and deficient tumour cells. Since BRCA2 is a pivotal protein in HR, it can be hypothesised that hyperthermic radiosensitisation is solely or at least mainly due to inhibition of HR. However, as reviewed earlier HT interferes with DNA repair pathways [Citation9,Citation18,Citation25–29]. Moreover, there is debate whether HT directly induces DNA breaks or not [Citation30,Citation31].
A very common technique to quantify the DNA DSBs is the γ-H2AX foci staining. Immediately after a DSB is formed, histone protein H2AX is phosphorylated by a kinase ATM (ataxia telangiectasia mutated). This phosphorylated histone H2AX (called γ-H2AX) recruits multiple DNA repair proteins to restore the DNA breaks. Remarkably, after HT alone, a slight induction of γ-H2AX foci can be found.
In this study, the impact of HT and PARP1-i on DNA repair mechanisms is investigated by subjecting BRCA2-proficient and deficient cells to different treatment combinations with ionising radiation, PARP1-i and HT. In particular, we studied the DNA DSBs by γ-H2AX, DNA damage by comet assay, cell survival by the clonogenic assay, cell cycle distribution by BrdU incorporation, apoptosis by the Nicoletti assay and DNA repair pathways protein levels by western blotting.
Material and methods
Cell culture
The mouse mammary cell lines KB2p1.21, KB2p3.4 (BRCA2-deficient) and KB2p1.21 R, KB2p3.4R3 (BRCA2-proficient) were established in the Netherlands Cancer Institute (NKI) as is described in [Citation32,Citation33]. The BRCA2-deficient cells were reconstituted with HSV-1/EBV amplicons which contained the intact mouse BRCA2 gene, resulting in the BRCA2-proficient cell lines [Citation34]. The reconstitute BRCA2-deficient cell lines, KB2p1.21 R and KB2p3.4R3 were purified by selection with 400 μg/ml hygromycin (Invitrogen). All cell lines were kindly provided by Jos Jonkers (NKI). These mammary cell lines were grown in DMEM F-12 (Life Technologies) supplemented with 10% heat-inactivated foetal bovine serum (FBS), 50 units/ml penicillin, 50 μg/ml streptomycin (Life Technologies), 5 μg/ml insulin (Sigma), 5 ng/ml epidermal growth factor (Life Technologies) and 5 ng/ml cholera toxin (Gentaur). γ-H2AX co-localisation to test the BRCA2-proficient cells on their functional BRCA2 reconstitution has been tested before on these cell lines [Citation32].
Reagents
Initial experiments were performed to find equitoxicity of PARP1-i (NU1025, Tocris Bioscience) on breast cancer cell lines, with different BRCA status. PARP1-i was dissolved in DMSO to treat cell lines with different single doses (0, 5, 10, 25, 50 and 100 μM). Equitoxic effects were determined and in the next experiments BRCA2-proficient cells were treated with a single treatment of 50 μM and as BRCA2-deficient cells were more sensitive to PARP1-i, they were treated with a dose of 25 μM. PARP1-i was added 30 min prior to HT and 90 min before ionising radiation for almost all experiments. For the comet assay, PARP1-i was added 72 h before HT and ionising radiation, for cells to have sufficient time to undergo cell division and incorporate the PARP1-i induced DNA damages.
Hyperthermia
Cells were treated with HT by partially submerging culture dishes in a thermostatically controlled water bath (Lauda aqualine AL12, Beun de Ronde, Abcoude, The Netherlands) for 1 h at 42 °C. Temperature check was performed in parallel dishes and the desired temperature (±0.1 °C) was reached in approximately 5 min. Cells were heated in a 5% CO2/95% air atmosphere with an air inflow of 2 l/min.
Radiation treatment
After PARP1-i and HT, cells were treated with multiple radiation doses (0, 2, 4, 6 and 8 Gy) to study clonogenic cell survival using gamma-rays from a 137Cs source at a dose rate of approximately 0.5 Gy/min. DNA damages monitored by the comet assay, apoptotic levels measured with the Nicoletti assay and cell cycle analyses were analysed after combinational treatment of PARP1-i, HT and a single treatment of 4 Gy. γ-H2AX foci was studied after combinational treatments of cells with a single dose of 2 Gy.
Immunohistochemistry
To study the effects of combinational treatments on the number of DSBs, γ-H2AX foci assay was conducted. Cells were seeded at a density of 3.0 × 105 cells on top of a sterile coverslip (21 × 26 mm) in a 6 cm culture dish. Approximately 2 h after cell seeding, medium was added to a total volume of 3 ml per dish. The next day, cells were treated with PARP1-i and/or HT and/or irradiated and again incubated at 37 °C. Cells were either fixed 30 min after treatment for homologous recombination activity (Rad51/γ-H2AX co-localisation) and for induction of DSBs (γ-H2AX only). To study remaining and irreparable DSBs, on the third day, approximately 24 h after irradiation, cells were washed with PBS before a 15-min fixation with 2% paraformaldehyde (PFA). After removing PFA with three times washing step of PBS, cells were permeabilised for 30 min with TNBS (PB containing 0.1% Triton X-100 and 1% FCS) at room temperature. Afterwards 50 μl per sample of the primary mouse monoclonal antibody anti-γ-H2AX (Millipore, diluted 1:100 in TNBS), or a cocktail of anti-γ-H2AX and RAD51 (dilution 1:100 and 1:25 respectively) was added under parafilm at room temperature, as described by Bergs et al. [Citation35]. Next, parafilm was removed and cells were washed once with PBS and twice with TNBS. Subsequently, a 30-min incubation with 50 μl per sample of the secondary goat-anti-mouse-Cy3 antibody (Jackson-Immunoresearch, diluted 1:100 in TNBS) covered with a parafilm, was performed. Again, at room temperature, but cells were kept in the dark. After removing the parafilm, cells were washed with TNBS, three times and finally to stain the nuclei, 50 μl of DAPI (2.5 μg/ml; Life technologies, USA) were added per sample for approximately 10 min. Eventually cells were washed once with PBS and coverslips were transferred upside down to microscope cover slides, before scoring foci under the fluorescence microscope (Leica). After washing, cells were incubated with TNBS for 30 min. Finally, vectashield with DAPI was dropped at the slide and the coverslip is turned upside down on the slide, for scoring under the fluorescence microscope. All experiments were performed in triplicate, analysing 100 cells per treatment.
Clonogenic assays
Cell survival was studied on BRCA2-proficient and BRCA2-deficient mammary cells lines after different treatment combinations; ionising radiation (0, 2, 4, 6 and 8 Gy), HT (1 h at 42 °C) and PARP1-i (50 μM for BRCA2-proficient cells and 25 μM in BRCA2-deficient cells). Clonogenic assays were performed as described by Franken et al. [Citation36]. First, cells were seeded 4 h prior to treatment into 6-well culture plates (Costar, USA). The 6-well plates were placed in an incubator with 5% CO2 at 37 °C until sufficiently large clones were formed (approximately 12–16 days). Afterwards, the medium was removed and cells were washed once with PBS. A 2–3 ml of a 6.0% glutaraldehyde and 0.5% crystal violet mixture was added for at least 30 min at room temperature. Next, dishes were washed with water and dried in normal air at room temperature. Colonies were counted under a light microscope [Citation37] and by dividing the plating efficiency of treated cells by that of control cells, surviving fractions were calculated [Citation38]. Survival curves were analysed using the linear-quadratic formula: S(D)/S(0) = exp − (αD + βD2) [Citation39]. S(D) is the survival at dose D and S(0) is the survival at dose 0.
Comet assay
The comet assay is a gel electrophoretic-based technique to explore DNA damage in individual cells. As DNA can get compromised by exposure to therapies, DNA breaks can occur. By permeabilising cells with a lysis buffer broken DNA ends can migrate out of the nucleus, and the negatively charged DNA breaks will migrate toward the anode during a 20-min electrophoresis. More damage will create a longer tail. Intact DNA is too large to migrate and therefore no or very minor tails may be observed. The comet assay was performed as in described by Olive and colleagues [Citation40]. First, 1% low-gelling-temperature agarose was mixed in distilled water, heated in a ∼100 °C water bath until it is completely dissolved (see-through). Next, the glass beaker is transferred to a 40 °C water bath to keep the agarose warm. Dust-free frosted-end microscope slides are pre-coated with this agarose mixture. About 500 μl of agarose is pipetted on the slide, covered with a coverslip (21 × 60 mm) and kept overnight in the fridge. The next day, after samples are treated, a 0.5% agarose mixture is prepared in the same way as before. After the agarose mixture is transferred to the 40 °C water bath, a cell pellet with approximately 50 000 cells is resuspended in 200 μl of agarose and pipetted on top of the pre-coated slide. Again, this is immediately covered with a coverslip (21 × 60 mm) and kept in the refrigerator for approximately 15 min. Next, coverslips are removed and cells are lysed in an alkaline lysis buffer (1.2 M NaCl, 100 mM Na2EDTA, 0.1% sodium lauryl sarcosinate, 0.26 M NaOH, pH >13) for 60 min in the fridge. Subsequently, microscope slides are transferred into an electrophoresis system and run for 20 min at 180 V. Finally, PI (2.5 μg/ml) was added on top of the slides before scoring cells under the fluorescence microscope (Leica).
Cell cycle analysis
Cell cycle distribution was studied by incorporation of the thymidine analogue 5-Bromo-2′-deoxy-uridine (BrdU, Sigma Aldrich, USA) in S-phase cells (60 min at 37 °C). A 1-h incubation at 37 °C with BrdU was performed 16 h after treatment and 1 h prior to fixation in 2 ml PBS and 6 ml of 100% ethanol. Next, cells were centrifugated at 1200 rpm for 5 min. Pellets were resuspended in pepsin-HCl (0.4 mg/ml, 0.1 N HCl) and cells were stored 30 min at room temperature. Cells were washed with PBT (0.5% Tween-20, Sigma Aldrich USA in 0.5 l PBS) and centrifugated afterwards. Pellets were resuspended in HCl (2 N, Merck) and cells were incubated 30 min at 37 °C Subsequently, cells were washed twice with PBS and once with PBTb (1% bovine serum albumin, Sigma, in PBT). After the last centrifugation, 100 μl primary antibody rat-anti-BrdU (Abcam, UK) in PBTb was added to the pellets for 1 h at 37 °C. Next, cells were washed twice with PBS and once with PBTg (1% normal goat serum, Dako, USA, in PBT). After the last centrifugation, 100 μl IgG goat-anti-rat FITC (Abcam, UK) in PBTg was added and left for 1 h at 37 °C. Again, cells were washed twice with PBS and finally, cells were resuspended in 500 μl and PI (2.5 μg/ml Sigma-Aldrich, USA) was added, cell suspensions were vortexed and directly measured using flow cytometry (FACS Canto, BD Biosciences, USA).
Apoptosis assay
To study apoptotic levels in cells after several combinational treatments, the Nicoletti assay [Citation41] was conducted in all cell lines to distinguish cells in different phases of the cell cycle including apoptosis. At 48 h after treatment cells, medium was collected in one 15 ml tube per treatment. Cells were washed with PBS and this was also collected in the same 15 ml tube. After trypsinisation, cells were neutralised with and resuspended in the medium of the tubes. Next, cells were centrifugated at 1200 rpm for 2 min. Eventually pellets were resuspended in Nicoletti buffer (0.1% w/v Sodium citrate, 0.1% v/v Triton X-100 in demi water, pH 7.4), before analysing samples using flow cytometry (FACS Canto, BD Biosciences, USA).
Western blotting
The effects of HT on HR or NHEJ were studied using western blotting. We examined the response to HT of BRCA2, a protein from the HR and ligase IV, a member of the NHEJ. Controls and treated cells were harvested 30 min after treatment. Pellets were lysed for 30 min on ice in ice-cold RIPA buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2EDTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) containing protein inhibitors [Citation42]. Laemmli buffer with 2-mercaptoethanol (355 mM) was added to the supernatant (1:1), before heated in boiling water for 2–5 min. Finally, samples were sonicated (Sonics & Materials Inc). One μg of protein was resolved by 4–15% SDS-PAGE gradient precast gels (BioRad) and transferred to PVDF membranes. Equal protein loading was checked by Ponceau S staining. Immunodetection was performed for anti-BRCA2 (antibodies-online), anti-ligase IV (abcam), anti-ERK2 (Bethyl Laboratories) and a secondary anti-rabbit (Invitrogen Life Technologies). All samples were enhanced using chemoluminescence (Amersham Pharmacia Biotech). Eventually, blots were analysed using LAS4000 (GE, Healthcare life sciences).
Statistics
All represented in vitro data have been performed at least three times, represented with ± standard error of the mean. SPSS (Chicago, IL, USA) was used to analyse tail length and apoptosis, using statistical software using a non-parametric Mann–Whitney test. Statistically significance differences in clonogenic assays were performed used the student t-test as this was normally distributed.
Results
BRCA2-deficient cells are sensitive to PARP1-i and HT
In both BRCA2-proficient and deficient cells, a few spontaneous DNA DSBs, using γ-H2AX foci staining were found in untreated cells (). Means ± SEM of three independently performed experiments are shown in the right panel of , demonstrating, HR is affected by HT in BRCA2-proficient cells. Ionizing radiation (2 Gy) increased the number of γ-H2AX foci. Unlike BRCA2-deficient cells, colocalisation of γ-H2AX and RAD51 was observed in the BRCA2-proficient cells. After combination of HT with ionising radiation, no colocalisation of γ-H2AX and RAD51 is observed.
Figure 1. Sensitivity of cells to PARP1-i and HT. (A) Left panel: In BRCA2-deficient cells, no accumulation of RAD51 is observed at sites of a double strand breaks after ionising radiation. In BRCA2-proficient cells, there is co-localisation of γ-H2AX (red) and RAD51 (green). Scale bar indicates 10 μm. Right panel: More than 100 cells per treatment, demonstrate that the homologous recombination in BRCA2-proficient cells is affected by HT. (B) To find an equitoxic effect of PARP1-i, cells are exposed to different doses of PARP1-i. From left to right: KB2p1.21 and KB2p3.4 (BRCA2-deficient cells), KB2p1.21 R and KB2p3.4R3 (BRCA2-proficient cells). An equitoxicity of ∼60% cell survival was found after treatment with 25 μM PARP1-i in BRCA2-deficient cells and 50 μM PARP1-i in BRCA2-proficient cells. Experiments were performed in duplicates. (C) In BRCA2-deficient cells (top panel) a lower cell survival is observed after addition of HT (42 °C for 1 h) to ionising radiation. Addition of PARP1-i (25 μM for BRCA2-deficient cells and 50 μM for BRCA2-proficient cells) to ionising radiation demonstrated a slightly lower cell kill compared to the combinational treatment of HT and ionising radiation. The BRCA2-proficient cell lines (bottom panel) showed some additional effect of PARP1-i to ionising radiation, while the effects of HT are more pronounced. The triple combination resulted in the highest cell kill. Means of at least three independent experiment with the standard error of the mean. p values: *p < 0.05.
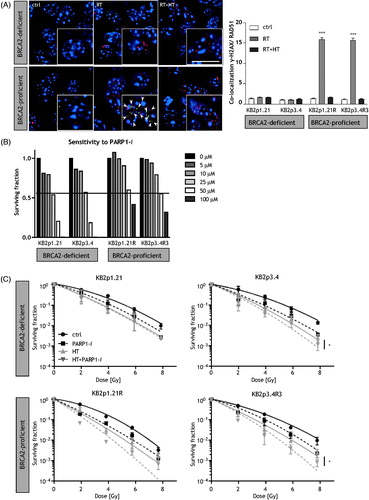
In order to determine the sensitivity, BRCA2-proficient and deficient cells were first treated with different doses of PARP1-i and cell survival was studied using clonogenic assay (). Consistent results were obtained in BRCA2-deficient cells (KB2p1.21 and KB2p3.4) and BRCA2-proficient cell lines (KB2p1.21 R and KB2p3.4R3). Approximately, 60% cell survival was observed after a single treatment of 25 μM and 50 μM PARP1-i in BRCA2-deficient and proficient cells lines respectively. These equitoxic concentrations were used in further experiments.
To explore whether BRCA2-deficient cells can be thermally radiosensitised, cells were exposed to combinational therapy of HT and ionising radiation. Cell survival was studied after cells were treated with ionising radiation alone or in combination with HT. demonstrates that both BRCA2-proficient and deficient cells have lower cell survival after combination with HT. The change in values of LQ-parameters, α and β, induced by HT treatment are of the same order of magnitude for both BRCA2-proficient and deficient cells. This indicates that radiosensitisation by HT is also significant in BRCA2-deficient cells wherein HR is disabled (). A 90-min incubation with PARP1-i prior to ionising radiation resulted in a lower cell survival in the BRCA2-proficient and deficient cell lines, with a larger effect in BRCA2-deficient KB2p3.4 versus KB2p3.4R3 cells. However, this is not observed when comparing KB2p1.21 versus KB2p1.21 R. The triple treatment with PARP1-i prior to ionising radiation and HT caused the most cell kill in all cell lines, especially in BRCA2-proficient cell lines.
Table 1. Linear-quadratic (LQ) parameters for cell survival after various treatments.
DNA damage in BRCA2-deficient cells is enhanced by HT
The comet assay was used to examine whether BRCA2-deficient cells have increased DNA damage after treatment with PARP1-i, HT and ionising radiation and to test if HT directly induced DNA damages or not (). In , representative comets of treated KB2p3.4 R cells are shown. Irradiation with 4 Gy highly increased the tail length, while HT alone did not induce any tail. Quantification of at least 100 cells per treatment are performed and illustrated in . Cells were treated with various combinations of HT (42 °C for 1 h), PARP1-i (at the doses obtained from ) and a single dose of 4 Gy (RT). In , nuclei are stained in blue and each red dot (γ-H2AX foci) represents one DSB. In the top lane, a single dose of 4 Gy radiation treated BRCA2-deficient cells show a 15–20 times longer tail length than the untreated cells. Interestingly, tail length did not significantly increase after HT (KB2p1.21 p = 0.10; KB2p3.4 p = 0.34; KB2p1.21 R p = 0.72; KB2p3.4R3 p = 0.07), but a 72-h incubation with PARP1-i did increase the tail length comparable to RT. Any combinational treatment of these cells with either RT or PARP1-i shows almost similar lengths of tails as RT or PARP1-i alone. In BRCA2-proficient cells (in the bottom lane) a single treatment with 4 Gy also demonstrated a tail, while HT alone does not. PARP1-i as a monotherapy induced shorter tails and the triple treatment induced the longest nuclear tails. Including HT with the combination of PARP1-i and RT was not significant in most cell lines (KB2p1.21 p = 0.46; KB2p3.4 p = 0.02; KB2p1.21 R p = 0.08; KB2p3.4R3 p = 0.00).
Figure 2. Higher induction of DNA damages in BRCA2-deficient cells after treatment with PARP1-i. (A) Different treatments have different effects on DNA damages. Representative cells of treated KB2P3.4 R are shown to demonstrate the effects of the comet assay. Scale bar indicates 10 μm. (B) Quantification of at least 100 cells was performed. Ionising radiation (RT) has a strong effect on induction DNA breaks, HT hardly induces any DNA breaks and PARP1-i is more effective in BRCA2-deficient cells than in BRCA2-proficient cells. Combinational treatments all have the ability to create DNA breaks, but the longest tails are observed after treatment including RT. Treatment with HT and PARP1-i has the most effects on BRCA2-deficient cells. (C) DNA DSBs can also be monitored with a γ-H2AX foci staining. Representative cells of treated KB2P1.21(R) illustrate the effects of various treatments, 24 h after treatment. (D) The bar graphs demonstrate the number of γ-H2AX foci, the induction of DSBs (30 min after treatment) and the remaining DSBs (24 h after treatment). γ-H2AX foci are counted for three independently performed experiments and per experiment a minimum of 100 cells were counted, which is presented by mean with the standard error of the mean. Scale bar indicates 5 μm. p values: *p < 0.05, **p < 0.01, ***p < 0.001.
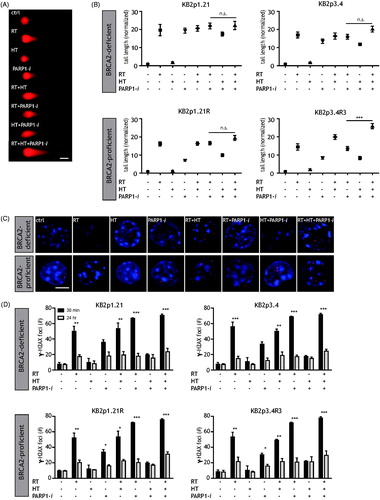
A 24-h time point was also studied by γ-H2AX foci staining to observe the unrepaired DNA DSBs (). In BRCA2 cells (a clear increase in γ-H2AX foci is observed after 2 Gy irradiation (). After HT only a minor increase in γ-H2AX foci is found, whereas treatment with PARP1-i demonstrated a high induction of γ-H2AX foci. The dual modality treatments with ionising radiation (RT and HT or RT and PARP1-i) demonstrated an even higher number of γ-H2AX foci, while the combination HT and PARP1-i showed less DSBs. The triple modality had the highest number of remaining γ-H2AX foci (24 h after treatment).
RT, HT and PARP1-i affect cell cycle arrest, apoptosis and DNA repair pathways
At 16 h post-treatment, cell cycle distribution was measured. In the untreated samples, a high percentage of cells are in the S-phase of the cell cycle (). Of all monotherapies, HT had the largest effect in inducing a G2-arrest. Even larger effects were observed for treatment with HT plus ionising radiation. The highest effect was seen after the triple modality in all cell lines.
Figure 3. Effects of RT, HT and PARP1-i on cell cycle progression, apoptosis and DNA repair pathways. (A) G2-arrest after addition of PARP1-i to HT and ionising radiation. Cell cycle was determined after BrdU incorporation using FACS analysis. HT and HT + RT and showed a clear increase in cells in G2-phase. Addition of PARP1-i to HT + RT increased the level of cells in G2-phase even more. (B) Induction of apoptosis after HT was most pronounced in BRCA2-proficient cells. Apoptotic levels were measured by the Nicoletti assay. Both RT and HT are able to induce increased apoptotic levels in BRCA2-deficient cells. However, PARP1-i hardly increased apoptosis in the BRCA2-deficient cells. The dual and triple modality all increased apoptotic levels. In BRCA2-proficient cells, considering the monotherapies, only HT had the ability to induce apoptosis. The dual modalities were all able to induce apoptosis, while the triple modality had het most pronounced increase in BRCA2-proficient cells. (C) Western blots demonstrating the effects of HR an NHEJ after HT. HT downregulates ligase IV (NHEJ) but not BRCA2 (HR) in BRCA2-deficient cells, and vice versa downregulates BRCA2 (HR) but not ligase IV (NHEJ) in BRCA2-proficient cells. p values: *p < 0.05, **p < 0.01, ***p < 0.001.
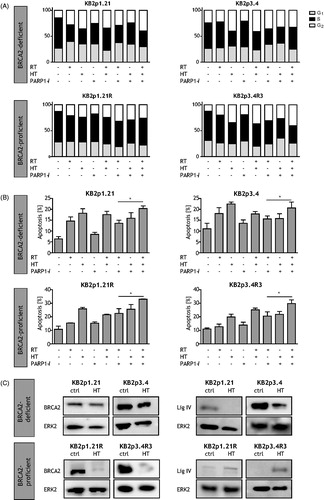
At 48 h post treatment, apoptotic levels were measured by flow cytometry. In , BRCA2-deficient cells (top lane) treated with 4 Gy, show increased apoptosis as compared to controls. Treatment with HT resulted in even higher apoptotic levels as compared to that of untreated cells. Even monotherapy with PARP1-i only resulted in a minor increase. Dual and triple modalities increased apoptotic levels in BRCA2-deficient cells. However, apoptotic levels were not higher than after HT alone. BRCA2-proficient cells (bottom lane) treated with RT hardly showed higher levels of apoptosis, while HT was effective in inducing apoptosis. Treatment of HT and RT in these cells and the triple modality resulted clearly in the highest levels of apoptosis. Addition of HT to the combination of PARP1-i and RT was significant in all cell lines (KB2p1.21 p = 0.003; KB2p3.4 p = 0.044; KB2p1.21 R p = 0.006; KB2p3.4R3 p = 0.018). This suggests that BRCA-deficient cells lines also benefit from HT.
Previous experiments demonstrated that BRCA2-deficient cells can also be sensitised by HT. Now we investigated whether HT also affects NHEJ, another important DNA repair pathway. In BRCA2-deficient cells lines, BRCA2 does not change after HT, while ligase IV (major protein of the NHEJ mechanism) levels are downregulated after HT (). In BRCA2-proficient cells after HT, BRCA2 is degraded, whereas ligase IV is upregulated. These results indicate that if cells have no functional HR pathway, cells compensate by using NHEJ, which can be vulnerable to HT.
Discussion and conclusions
When PARP1 is inhibited, the integrity of DNA is salvaged by HR. However, when HR is blocked by HT DNA damage accumulates resulting in higher cell kill. With the experiments performed in this study we aimed to treat cells with a dose of PARP1-i that caused equitoxicity in both BRCA2-proficient and deficient cell lines. The equitoxic dose of PARP1-i yielded similar cell survival fractions and induced radiosensitisation in BRCA cell lines. Due to the equitoxic PARP1-i doses, BRCA2-deficient cell lines were as much sensitive to combined treatment of PARP1-i with ionising radiation as BRCA2-proficient cell lines (). Furthermore, the hyperthermic radiosensitisation also had a significant effect on BRCA2-deficient cell lines, indicating that these cells are sensitive to HT as well. Since HR is not active in BRCA2-deficient cell lines, the hypothesis of considering HT induced radiosensitisation being solely due to HR does not hold true. This paves way to the possibility of the involvement of another molecular mechanism.
As can be observed in , at all conditions the value of the α parameter is enhanced. This is important for the clinical application of the treatments. Hyperthermia and/or PARP1 inhibition radio-enhance both BRCA2-deficient and BRCA2-proficient cells. This indicates that many different mechanisms are involved. Hyperthermia can affect HR and consequently improve radiation treatment. PARP1 inhibition restrains SSB repair which can result in radiosensitisation. The triple modality did not result in a lower survival fraction of KB2p1.21 cells compared to the dual modalities, although in the KB2p3.4 cells there seems to be a slightly additional effect of PARP1 inhibition. In BRCA2-proficient cell lines, radiosensitisation by PARP1-i and HT had almost equal effects, but triple modality definitely showed a lower cell survival.
DNA damage measured by the comet assay, demonstrated long tails after ionising radiation, indicating the presence of high levels of DNA DSBs (). Treatment with PARP1-i alone, exhibited a pronounced DNA damage in BRCA2-deficient cells than in BRCA2-proficient cells. These findings confirm previously published results [Citation33].
What did our present results add to the controversy whether HT directly induces DNA breaks [Citation30,Citation43] or not [Citation9,Citation44]? We found that HT alone does not induce a tail in the comet assay, or at the most a very minor one. This suggests that HT does not directly, induce DNA DSBs. Only in the BRCA2-proficient cell lines, the triple modality led to a higher number of DNA DSBs. In the KB2p1.21, no significant difference was observed between dual modalities and the triple modality. Thus, including HT to PARP1-i and RT or supplementing PARP1-i to the combination of RT + HT did not increase the induction of DNA DSBs. However, in KB2p3.4, there is a slight increase in induction of DNA DSBs compared to dual modalities. Even 24 h after treatment a significantly high number of DNA DSBs (γ-H2AX foci) were observed (). Likewise, these experiments provide more evidence that HT does not directly induce high levels of major DNA damage.
HT is the only monotherapy that has the ability to highly increase apoptotic levels () and lead to a higher percentage of cells in G2-arrest (). In combination with ionising radiation, these effects were more pronounced after the triple combination. The G2-arrest is elevated in the KB2p3.4 cells compared to the KB2P1.21 cells.
Using γ-H2AX and RAD51 foci staining, we confirmed that BRCA2-deficient cells do not have functional HR, as a single dose of 2 Gy did not cause colocalisation of γ-H2AX and RAD51 in BRCA-deficient cells, while this did occur in BRCA2-proficient cells. After HT, colocalisation disappeared in BRCA2-proficient cells, as HT inhibits the HR, and consequently RAD51 is not accumulated at sites of DNA DSBs [Citation18,Citation35,Citation45]. There seem to be contradictory results on the influences of HT on NHEJ. Whereas several publications demonstrate that NHEJ is upregulated when HR is inhibited by HT [Citation35,Citation46], others suggest that HT downregulates the NHEJ [Citation47,Citation48] and in some cases no differences were found in NHEJ [Citation49]. Moreover, several groups claim that the hyperthermic effects on NHEJ do not play a crucial role in enhancement of thermal radiosensitivity [Citation48,Citation49].
According to previous findings and our recent results, it seems that the effects of HT are cell line and temperature dependent whether the effects on NHEJ are noticeably, up- or down-regulated. Our results demonstrate that BRCA2-deficient cells can also be sensitised by HT and affect the NHEJ protein ligase IV in both the BRCA2-deficient and proficient cells. In the BRCA2-deficient cells lines, BRCA2 does not change after HT—as this protein is not functional, while the ligase IV levels are reduced after HT. Perhaps, because these cells rely completely on NHEJ, effects are noticed within this pathway. In BRCA2-proficient cells, on the other hand, BRCA2 is downregulated, while ligase IV is upregulated. Since the BRCA2-proficient cells lines have active HR and NHEJ in untreated samples, when HT downregulates HR, cells that need to restore DNA damage will chose the NHEJ, causing an increase in ligase IV.
Overall, as PARP1 levels are increased after ionising radiation induced DNA damages [Citation50,Citation51], we have shown that PARP1-i is capable of increasing the DNA damages, resulting in reduction of cell survival. Also in BRCA2-proficient cells lines, treatment with PARP1-i resulted in a lower surviving fraction.
Previous studies demonstrated the selective sensitivity of BRCA2-mutated tumours to PARP1-i [Citation52,Citation53]. Here, we demonstrate that BRCA2-deficient cells are also sensitive to HT, resulting in higher levels of unrepaired ionising radiation induced DNA damages, resulting in higher cell kill. The BRCA2-proficient cells treated with RT, HT and PARP1-i show higher values of the linear parameter α when compared to BRCA2-deficient cells treated with RT and PARP1-i. These results suggest that cancer patients who are non-BRCA2 carriers would benefit if PARP1-i are combined with radiotherapy and HT. Furthermore, we also demonstrate that patients without a functional BRCA2 will have advantage of a treatment including HT. The next step towards clinical investigations requires in vivo confirmation of effectivity and investigation of normal tissue toxicity.
Acknowledgement
The authors thank Jos Jonkers for providing us with BRCA2 cell lines (KB2p1.21, KB2p1.21 R, KB2p3.4 and KB2p3.4R3). This work was supported by the Dutch Cancer Society (UVA 2012–5540).
Disclosure statement
The authors report no declarations of interest
Additional information
Funding
References
- Branzei D, Foiani M. (2010). Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11:208–19.
- Satoh MS, Lindahl T. (1992). Role of poly(ADP-ribose) formation in DNA repair. Nature 356:356–8.
- Shrivastav M, De Haro LP, Nickoloff JA. (2008). Regulation of DNA double-strand break repair pathway choice. Cell Res 18:134–47.
- Essers J, van Steeg H, de Wit J, et al. (2000). Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J 19:1703–10.
- Rothstein R, Michel B, Gangloff S. (2000). Replication fork pausing and recombination or "gimme a break". Genes Dev 14:1–10.
- Bryant HE, Schultz N, Thomas HD, et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913–17.
- Farmer H, McCabe N, Lord CJ, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917–21.
- Eppink B, Krawczyk PM, Stap J, Kanaar R. (2012). Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. Int J Hyperthermia 28:509–17.
- Oei AL, Vriend LE, Crezee J, et al. (2015). Effects of hyperthermia on DNA repair pathways: one treatment to inhibit them all. Radiat Oncol 10:165.
- Falk MH, Issels RD. (2001). Hyperthermia in oncology. Int J Hyperthermia 17:1–18.
- Oleson JR, Dewhirst MW. (1983). Hyperthermia: an overview of current progress and problems. Curr Problems Cancer 8:1–62.
- Horsman MR, Overgaard J. (2007). Hyperthermia: a potent enhancer of radiotherapy. Clin Oncol (R Coll Radiol) 19:418–26.
- Oei AL, van Leeuwen CM, ten Cate R, et al. (2015). Hyperthermia selectively targets human papillomavirus in cervical tumors via p53-dependent apoptosis. Cancer Res 75:5120–9.
- Repasky EA, Evans SS, Dewhirst MW. (2013). Temperature matters! And why it should matter to tumor immunologists. Cancer Immunol Res 1:210–16.
- Dewhirst MW, Lee CT, Ashcraft KA. (2016). The future of biology in driving the field of hyperthermia. Int J Hyperthermia 32:4–13.
- Horsman MR. (2016). Realistic biological approaches for improving thermoradiotherapy. Int J Hyperthermia 32:14–22.
- Crezee H, van Leeuwen CM, Oei AL, et al. (2016). Thermoradiotherapy planning: Integration in routine clinical practice. Int J Hyperthermia 32:41–9.
- Krawczyk PM, Eppink B, Essers J, et al. (2011). Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci USA 108:9851–6.
- El-Awady RA, Dikomey E, Dahm-Daphi J. (2001). Heat effects on DNA repair after ionising radiation: hyperthermia commonly increases the number of non-repaired double-strand breaks and structural rearrangements. Nucleic Acids Res 29:1960–6.
- van der Zee J, Gonzalez Gonzalez D, van Rhoon GC, et al. (2000). Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: a prospective, randomised, multicentre trial. Dutch Deep Hyperthermia Group. Lancet 355:1119–25.
- Datta NR, Rogers S, Klingbiel D, et al. (2016). Hyperthermia and radiotherapy with or without chemotherapy in locally advanced cervical cancer: a systematic review with conventional and network meta-analyses. Int J Hyperthermia 32:809–21.
- Snider JW III, Datta NR, Vujaskovic Z. (2016). Hyperthermia and radiotherapy in bladder cancer. Int J Hyperthermia 32:398–406.
- Datta NR, Puric E, Klingbiel D, et al. (2016). Hyperthermia and radiation therapy in locoregional recurrent breast cancers: a systematic review and meta-analysis. Int J Radiat Oncol Biol Phys 94:1073–87.
- Issels RD. (2008). Hyperthermia adds to chemotherapy. Eur J Cancer 44:2546–54.
- Raaphorst GP, Freeman ML, Dewey WC. (1979). Radiosensitivity and recovery from radiation damage in cultured CHO cells exposed to hyperthermia at 42.5 or 45.5 degrees C. Radiat Res 79:390–402.
- Freeman ML, Raaphorst GP, Dewey WC. (1979). The relationship of heat killing and thermal radiosensitization to the duration of heating at 42 degrees C. Radiat Res 78:172–5.
- Dewey WC. (1996). Tumour reoxygenation and response after hyperthermia and radiation: T(90) as a predictor and T(50) as the cause?. Int J Hyperthermia 12:443–4.
- Iliakis G, Wu W, Wang M. (2008). DNA double strand break repair inhibition as a cause of heat radiosensitization: re-evaluation considering backup pathways of NHEJ. Int J Hyperthermia 24:17–29.
- Corry PM, Robinson S, Getz S. (1977). Hyperthermic effects on DNA repair mechanisms. Radiology 123:475–82.
- Kampinga HH, Laszlo A. (2005). DNA double strand breaks do not play a role in heat-induced cell killing. Cancer Res 65:10632–3.
- Velichko AK, Petrova NV, Kantidze OL, Razin SV. (2012). Dual effect of heat shock on DNA replication and genome integrity. Mol Biol Cell 23:3450–60.
- Evers B, Drost R, Schut E, et al. (2008). Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res 14:3916–25.
- Evers B, Schut E, van der Burg E, et al. (2010). A high-throughput pharmaceutical screen identifies compounds with specific toxicity against BRCA2-deficient tumors. Clin Cancer Res 16:99–108.
- Wade-Martins R, Smith ER, Tyminski E, et al. (2001). An infectious transfer and expression system for genomic DNA loci in human and mouse cells. Nat Biotechnol. 19:1067–70.
- Bergs JW, Krawczyk PM, Borovski T, et al. (2013). Inhibition of homologous recombination by hyperthermia shunts early double strand break repair to non-homologous end-joining. DNA Repair 12:38–45.
- Franken NA, Rodermond HM, Stap J, et al. (2006). Clonogenic assay of cells in vitro. Nat Protoc 1:2315–19.
- Bergs JW, Franken NA, ten Cate R, et al. (2006). Effects of cisplatin and gamma-irradiation on cell survival, the induction of chromosomal aberrations and apoptosis in SW-1573 cells. Mutat Res 594:148–54.
- van Bree C, Franken NA, Snel FA, et al. (2001). Wild-type p53-function is not required for hyperthermia-enhanced cytotoxicity of cisplatin. Int J Hyperthermia 17:337–46.
- Barendsen GW. (1997). Parameters of linear-quadratic radiation dose-effect relationships: dependence on LET and mechanisms of reproductive cell death. Int J Radiat Biol 71:649–55.
- Olive PL, Banath JP. (2006). The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1:23–9.
- Riccardi C, Nicoletti I. (2006). Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 1:1458–61.
- van Bree C, Franken NA, Rodermond HM, et al. (2004). Repair of potentially lethal damage does not depend on functional TP53 in human glioblastoma cells. Radiat Res 161:511–16.
- Takahashi A, Mori E, Nakagawa Y, et al. (2017). Homologous recombination preferentially repairs heat-induced breaks in mammalian cells. Int J Hyperthermia 33:336–42.
- Hunt CR, Pandita RK, Laszlo A, et al. (2007). Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res 67:3010–17.
- Genet SC, Fujii Y, Maeda J, et al. (2013). Hyperthermia inhibits homologous recombination repair and sensitizes cells to ionizing radiation in a time- and temperature-dependent manner. JCell Physiol 228:1473–81.
- van Oorschot B, Granata GD, Franco S, Ten Cate R, Rodermond HM, Todaro M, et al. (2016). Targeting DNA double strand break repair with hyperthermia and DNA-PKCS inhibition to enhance the effect of radiation treatment. Oncotarget 7:65504–13.
- Ihara M, Takeshita S, Okaichi K, et al. (2014). Heat exposure enhances radiosensitivity by depressing DNA-PK kinase activity during double strand break repair. Int J Hyperthermia: 30:102–9.
- Kampinga HH, Dynlacht JR, Dikomey E. (2004). Mechanism of radiosensitization by hyperthermia (> or =43 degrees C) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia 20:131–9.
- Woudstra EC, Konings AW, Jeggo PA, Kampinga HH. (1999). Role of DNA-PK subunits in radiosensitization by hyperthermia. Radiat Res 152:214–18.
- Schiewer MJ, Goodwin JF, Han S, et al. (2012). Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov 2:1134–49.
- Zaremba T, Thomas HD, Cole M, et al. (2011). Poly(ADP-ribose) polymerase-1 (PARP-1) pharmacogenetics, activity and expression analysis in cancer patients and healthy volunteers. Biochem J 436:671–9.
- Tinker AV, Gelmon K. (2012). The role of PARP inhibitors in the treatment of ovarian carcinomas. Curr Pharm Des 18:3770–4.
- Bangham M, Goldstein R, Walton H, Ledermann JA. (2016). Olaparib treatment for BRCA-mutant ovarian cancer with leptomeningeal disease. Gynecol Oncol Rep 18:22–4.