Abstract
A PUF-ICP-MS method has been developed that can quantify a large number of elements in size-resolved ambient aerosol samples collected over short-time intervals (four hours in this study). The pre-cleaning and digestion protocols developed enabled the quantification of 29 elements (including Hg) in ambient PM sampled onto a PUF substrate. The microwave-assisted acid digestion method affects a total dissolution of both aerosol and substrate, producing a colorless solution with no trace of aerosol, PUF, or polypropylene particulates, and is compatible with many analytical techniques (e.g., ICP-MS, CVAF, GFAA). For the large majority of elements the PUF-ICP-MS method provides superior elemental detection capabilities than traditional XRF approaches and importantly, method detection limits (MDLs) for most studied elements are lower than achieved with a 4-hour MOUDI approach using optimized ICP-MS analysis. For example, MDLs for V, Sr, Cd, Sb, and Hg are respectively 0.15, 0.18, 0.034, 0.051, and 0.0055 (solvent cleaning only for Hg) ng m−3. Only one-third, or less, of the PUF substrate is required for the ICP-MS analysis leaving the majority of the substrate available for additional characterization, e.g., organic speciation, of the same air mass. A demonstration study conducted in Phoenix, Arizona, shows that this new method can quantitatively resolve differences in element concentrations on short time scales and has the potential to be a powerful new tool for identifying sources of trace and ultra-trace elements. The highly size and time resolved data can also provide useful information for the study of health effects of individual or groups of elements.
INTRODUCTION
Particulate matter (PM) is a subject of intense study not only due to climate forcing and visibility concerns but also because of known and suspected health effects. Concentrations of PM below 2.5 μ m in aerodynamic diameter (PM2.5) are regulated in the United States to protect human health and there is mounting evidence that ultrafine particles (PM below 100 nm in aerodynamic diameter) are more hazardous to human health than larger particles (CitationOberdorster 2001). Though much work remains in identifying the components of PM responsible for health impacts, many heavy metals have been shown to be toxic (CitationLingard et al. 2005; CitationRiley et al. 2003) and their emissions are regulated by the USEPA (National Emission Standards for Hazardous Air Pollutants) and by states (for example, California's Air Toxic Program). Trace elements, including most metals, released by high temperature anthropogenic sources are principally emitted in the submicrometer particle size range (CitationOndov and Wexler 1998). Coarse particle modes are also significant components of the total aerosol for many elements, reflecting contributions from soils and dust, and therefore improved source attribution depends in part on obtaining size-resolved data. However, concentrations of metals are typically low (CitationOndov and Wexler 1998) requiring use of very sensitive methods for accurate measurement, and this requirement is further stressed as aerosol populations are fractionated (CitationCarlton et al. 1999; CitationUSEPA 1999; CitationWang et al. 1995a). Also driving the need for novel sampling and analytical methods that can quantify at even lower concentrations are recent concerns about acute health effects resulting from exposures of less than 24 hours (CitationPeters et al. 2001).
PM2.5 concentrations of individual elements range from the order of μg m−3 for common elements such as aluminum and calcium to pg m−3 for ultra-trace metals such as cadmium and uranium. Inductively Coupled Plasma Mass Spectrometry-based (ICP-MS) methods have been used on 24-hour PM samples collected on Teflon substrates to measure concentrations of a large suite of elements in PM2.5 and size fractions below 1.8 μm (CitationDillner et al. 2005), however traditional sampling methods, even when coupled with extremely sensitive analytical techniques such as ICP-MS, are inadequate to capture short time-scale cycles of trace elements in ambient PM. In this study, a method of pre-cleaning and analyzing PM collected on polyurethane foam (PUF) substrates was developed. This novel analytical approach was paired with high-volume air sampling to provide size-segregated concentrations of a very large set of elements on short time scales. This new method will support detailed chemical and toxicological studies of PM that are difficult or impossible to implement by traditional approaches.
METHODS
Sampling
To obtain high time resolution size-segregated PM samples, a Rupprecht and Patashnick, ChemVol Model 2400 High Volume Cascade Impactor (CitationDemokritou et al. 2002) with six sampling stages was used. The sampler was operated at the manufacturer recommended flow rate of 760 liters per minute (lpm) and based on unpublished data from the manufacturer for the stages used, particles were collected in the following diameters size ranges, < 0.1, 0.1–0.5, 0.5–1.0, 1.0–2.5, 2.5–10, > 10 μm. The PUF substrates used in this sampler enable high flow rates and mass loadings to be achieved. In addition, the PUF substrates exhibit high particle collection efficiency with minimal bounce. Micro-orifice, uniform deposit (MOUDI) samplers, which are traditionally used to obtain size segregated samples, operate at 30 lpm making short time scale analysis of trace elements difficult, even for highly polluted air urban areas. In addition, the Teflon and foil impaction substrates used in the MOUDI are more prone to particle bounce than the polyurethane foam substrates used in the ChemVol.
The PUF impactor substrates employed in the ChemVol 2400 sampler are toroid-shaped with a thickness (depth) of 6.4 mm and OD/ID of 165.1/88.9 mm, 139.7/114.3 mm, and 133.4/120.6 mm for the PM10, PM2.5, and three stages below PM2.5, respectively. PUF weights range from 0.5 grams for the sub-micron stages to ∼ 2 grams for the PM10 stage substrate. The accelerator slits immediately upstream of the PUF in each stage produce narrow (several mm width) PM deposits on the PUF (CitationDemokritou et al. 2002). Three identically sized slits at each stage deposit PM on ∼ 85% of the PUF substrate. PM mass loadings on the PUF substrates will obviously depend upon ambient PM levels, sampling duration, and size distribution, but as a case in point, sampling for 4 hours where the ambient PM level is 20 μ g L−1, with 40% of PM mass in the fines, would result in a total of 1.5 mg of PM on the PM2.5 PUF stage. This loading translates to ∼ 40 μ g PM per cm (2.5 μ g per mg) of PUF.
To test the viability of this method, two sets of samples were collected on March 25, 2003, one in the morning (6 A.M. to 10 A.M.) and the other during midday (11 A.M. to 3 P.M.) in Phoenix, Arizona. The sampling site was located approximately 4 miles north of downtown Phoenix, one mile east of Interstate 17 and 3 miles west of Highway 51 and 3 miles north of Interstate 10, all major arteries in the city. During the sampling periods, the wind was coming from the north-northeast during the morning at a steady 0.3 meters per second (m s−1) and east-northeast during midday ranging in speed from 0.8 to 1.6 m s−1, providing regional aerosol from the Phoenix area, not aerosol dominated by any of the major highways.
PUF Preparation
The PUF (upper 5-size fractions) and polypropylene (after-filter, < 0.1 μ m size cut) substrates were cleaned prior to use to minimize elemental and organic background. The PUF application/preparation described herein is intended to be compatible with a range of analytes; organics, and polyatomic ions, in addition to trace elements. Solvents are required for the organic application (and in general do not compromise the elemental application) and in selecting options for trace element removal, acids were considered, but ultimately a detergent was chosen for the following reasons. (1) A detergent improves the blanks for both organic and most inorganic analytes; (2) Residual acid is a major concern for polyatomic ion application—it's nearly impossible to remove nitrate and chloride from an acid cleaned substrate (especially from PUF) to levels necessary for ambient sampling; and (3) Deterioration of the PUF matrix upon acid exposure (particularly if PUFs were not immediately used), leading to particle generation and collection efficiency variations, is unacceptable.
Substrate cleaning was performed in a pre-cleaned, wide mouth, 2 L glass jar with a Teflon-lined polypropylene cap.
| a. | 1 L of 2% Alconox solution was added, the jar shaken for 2 minutes, the substrates allowed to soak for 30 minutes, the jar shaken for 2 more minutes, and the detergent solution then poured off. | ||||
| b. | 1 L of MQ-water was added, the jar shaken for 2 minutes, and the rinse water discarded. The water rinse step was repeated an additional 6 times. | ||||
| c. | The substrates were placed onto a Pyrex glass dish and then inserted into a vacuum desiccator for 6 hours to dry. | ||||
| d. | Substrates were then sonicated [Branson model B-3200 (40 kHz), 30°C] in isopropanol for 20 minutes. | ||||
| e. | Substrates were transferred to dichloromethane and sonicated for an additional 20 minutes. | ||||
| f. | Finally, the substrates were dried in a vacuum desiccator. | ||||
This article specifically addresses the inorganic PUF application. A comparison of element levels in PUF substrates cleaned using the protocol outlined above with a shortened cleaning protocol using just the organic solvents showed that the full procedure produced lower (11 elements) or equivalent (8 elements) blanks (3 elements were below detection, so comparisons could not be made). This was particularly evident for most of the low molecular weight “major elements” (Ca, Al, Fe, K, Mg). However for 7 elements (Na, Ba, Cu, Cr, Rb, Y, Hg), likely sourced from the Alconox, the solvent-only cleaning method produced the best PUF blanks. Alconox is 20–30% by weight Na; and Ba, Y, and Cr are common contaminants in the chemical components of Alconox. Tin levels in the PUF material (added as a stabilizer during manufacturing) were exceptionally high (270 μ g g−1) and not significantly lowered by cleaning—therefore tin concentrations in PM could not be reported. The high levels of tin produced a large, poorly corrected isobar on the isotope (114) of cadmium most often used in ICP-MS analysis—we used the Cd-111 isotope instead. Platinum levels in the PUF material were also relatively high (0.035 μ g g−1) possibly the result of its use as a catalyst/curing agent in the production of polyurethane foam. Concentrations of most other elements in the cleaned PUF () were lowered to levels that did not compromise detection of that element in ambient PM. A parallel evaluation of the efficacy of this method for PM-associated Hg was performed (a cold-vapor atomic fluorescence [CVAF] analysis technique was applied for Hg, not the ICP-MS method used for all other elements). The solvent-only cleaning method produced lower and less variable blanks than the full procedure (again, likely attributable to the Alconox), and this PUF blank represented only a very small (< 4%) fraction of the total PM-associated Hg collected in a typical 4-hour sampling period. The PUF digest presented few analytical difficulties for the CVAF technique—therefore we conclude that the overall method shows great promise for determining the size distribution of aerosol-associated mercury.
TABLE 1 Concentrations (μg−1) of target analytes in PUF substrates after full cleaning and solvent-only cleaning. Mean ± 1 standard deviation, n = 6.
After sampling the substrates are transferred to pre-cleaned amber glass jars fitted with Teflon-lined caps and stored at −20°C.
ELEMENT ANALYSIS
After dissolution of the PM (and substrates), ICP-MS was used to determine the total concentrations of 28 elements. The microwave-assisted dissolution procedure we developed for the PM/substrates was designed to achieve not only a quantitative solubilization of the PM, but also of the substrates. Our group and others (CitationSmith and Arsenault 1996; CitationSwami et al. 2001; CitationWang et al. 1995b; CitationYang et al. 2002) have demonstrated that microwave-based techniques are robust and powerful tools for PM solubilization. We pursued this approach to ensure complete recovery of all embedded particulates (and associated elements). Methods that attempt to remove PM from substrates risk both poor recovery and contamination. Analytical methods that are based on direct solids analysis of the PUF/particles (e.g., XRF, PIXIE, INAA) are generally limited in sensitivity and matrix effects can be problematic (CitationGraney et al. 2004; CitationPyle et al. 1996; CitationUSEPA 1999). Sample handling and analysis was performed using trace metal clean techniques. All preparation steps and analyses were preformed in HEPA filtered, dedicated trace metal clean laboratories by personnel with extensive experience in trace level techniques. Substrates were handled with Teflon forceps in polyethylene-gloved hands.
Digestion
PUF and polypropylene substrates were sectioned under a Class 100 HEPA hood, using ceramic blades. Typically, replicate 50 mg sections were prepared. PUF masses greater than this resulted in over-pressurization of the microwave bombs. A microwave-assisted acid digestion, in micro (6-ml)-Teflon bombs, was used to effect a total dissolution of both aerosol particles and substrate. The resulting solution is colorless with no trace of aerosol, PUF or polypropylene particulates. To minimize organic-loading to the microwave vessels, a 30-hour pre-digestion at room temperature using nitric acid (1.5 mL of 16N HNO3) and hydrogen peroxide (0.5 mL of 30% H2O2) was performed in the open bomb before microwave solubilization. To complete the digestion, 0.38 mL of 12 N HCl and 0.10 mL of HF were added to the bomb, the vessel sealed and the microwave program initiated. A Milestone Ethos+ temperature-controlled automated microwave system (1000 W) was used with a program that slowly ramped the temperature to 180°C with intermediate holds at both 140°C (6-minute ramp, 2-minute hold) and 165°C (5-minute ramp, 6-minute hold). A 4-minute ramp and 15-minute hold at 180°C completed the microwave program. After cooling, the bomb contents were quantitatively transferred to trace metal clean LDPE bottles and diluted to 30.0 mL. All acids used were of ultra-high purity grade (Baker Ultrex® or Fisher Optima®). Each batch of 16 samples was accompanied by three method blanks, two spikes, and two samples each of three different NIST Standard Reference Materials (STM, Recycled Auto Catalyst (#2556), Urban Dust (#1649a), and San Joaquin Soil (#2709)) to monitor blanks and analyte recoveries. Representative pre-cleaned substrate blanks were also incorporated as samples in the digest batch.
ICP-MS Analysis
The concentrations of 28 elements (Ca, Al, Fe, K, Mg, Na, Ti, Ba, Zn, Cu, Mn, Sr, Pb, Sb, Cr, Rb, Ce, Mo, V, W, Y, Cs, Rh, Pd, Ag, U, Cd, Pt) in the digests were determined by ICP-MS. A Thermo PQ-ExCell ICP-MS interfaced with a Cetac microconcentric desolvating nebulizer (MCN-6000) was employed. The later provided an inert all Teflon introduction system that exhibits good washout characteristics and very low blanks. The desolvating MCN significantly reduced oxide and chloride interferences, and with an uptake rate of 100 μ L/minute, allowed for micro-sample volumes. To eliminate molecular isobars that compromise the analysis of an important group of 6 light elements when traditional ICP-MS is applied, the “cool”-plasma, shielded torch mode of the ExCell ICP-MS was utilized for Al, Ca, Fe, K, Mg, and Na. With the MCN-6000 front-end the ICP-MS sensitivity was in the range of 80–100,000 cps/ppb with backgrounds typically in the range of 0.5 to 2 cps. Instrumental detection limits (3× the standard deviation of dilute acid blanks) were in the low ng L−1 range. Metal quantification was performed using external standards (High Purity Standards, Charleston SC) with internal normalization (using Ga, In, Bi as internal standards). Isotopes were acquired in peak jumping mode with a minimum of four replicate 3–7-minute analyses (60 sweeps, 10 ms dwell/isotope) performed on each sample after a 60-second uptake and stabilization period. It was determined that to achieve low to sub-nanogram per liter quantification throughout the analytical batch sequence an extended rinse (240 seconds) with 2% high purity nitric acid was necessary to prevent trace carry-over of certain “sticky” elements (Ag, Pd, Sn) from calibration and check standards and Standard Reference Materials (SRMs, whose concentrations could be several orders of magnitude greater than trace level samples) distributed throughout the batch. This approach also accommodated ultra-trace quantification from the wide range of sample concentrations, and more efficiently reconditioned the ICP-MS cones between samples. The small loss in productivity resulting from the extended rinse time was more than compensated by the performance enhancement.
The mean concentration and standard deviation of the four analyses was determined. Occasionally, using a standard Q-test, one of the four data points was judged to be an outlier, in which case the outlier was excluded from the summary statistics. Blank concentrations were calculated as the mean of three batch specific matrix blanks or appropriate field method blank. The concentration of an element in each method blank was determined four times and also subjected to a Q-test. If one measurement was determined to be an outlier, that data point was excluded from the mean. Silicon is not determined as Si is partially lost as volatile SiF4 under the digestion conditions employed. If silicon data is required, a similar acid digestion can be carried-out in sealed 6 mL Savillex Teflon vials, with quantitative recovery of silicon. Ni data was not acquired due to elevated instrument background. Interference problems in the ICP-MS analysis (unrelated to the PUF matrix or digestion protocol) resulted in relatively large uncertainties in the As and Se data. Therefore, these data are not shown. Arsenic concentrations in the PUF are ∼ 0.04 micrograms per gram (∼ 0.02 ng m− 3 under conditions outlined in ).
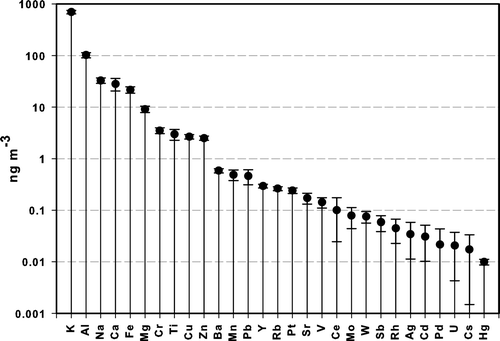
FIG. 1 PUF blank equivalent concentrations based on a 180 m3 air sample (4-hour collection at nominal 760 L min−1 sampling rate). The values shown are the average of blanks from two size cuts and the uncertainty represents ± 1 standard deviation.
Data Analysis
Reported element concentrations were method (PUF/polypropylene) blank corrected with all uncertainty estimates propagated. Because the mass of PUF sample and blank sub-samples digested varied slightly, blank corrections were performed on a per unit PUF mass basis, that is, ((sample PUF element mass/sample PUF mass)–(blank PUF element mass/blank PUF mass)) × sample PUF mass = blank corrected element mass per PUF section sampled. Where possible, PUF field blanks from the same size cut were matched with samples from that size cut. Propagated (sum-of-squares) uncertainty components included in this ICP-MS–derived element mass value included sample analysis precision and blank analysis precision. Air concentrations of each element were then determined by dividing the corrected element mass per PUF section by the volume of air sampled by that section. An additional uncertainty of 5% was propagated into the air concentration values to reflect error associated with the air volume measurement.
To establish the uniformity of deposition of PM on the PUF and assess overall precision of the method, we analyzed four separate sub-samples of the same loaded PUF substrate. A remarkable consistency was observed with the precision (RSD) of most of the 30+ elements examined in the 3–7% range. A few elements exhibited greater variability, and included those negatively impacted by the full cleaning (Na-36%, Cr-21%), and those present near the method LOD (Pd-95%, W-62%), and Mo-26% and Fe-38%.
RESULTS
Method Development
PUF blank equivalent concentrations for the 29 elements are shown in . Blanks presented in are for a 4-hour (180 m3) collection period to correspond with the sampling interval used for field data collection. PUF blanks for 19 of the 29 elements are less than 1 ng m−3, and 11 of these are below 0.1 ng m−3. Mercury PUF blank equivalent concentrations (data from cleaning trials) averaged ≤0.01 ng m−3 (0.0097 ng m−3 solvent-only cleaning). Higher blank levels were observed for many of the “major” elements; however the sample mass loading of these elements typically was high enough to minimize the blank contribution.
Concentrations of many elements in the polypropylene after-filter (< 0.1 μm fraction) blanks were similar to that of the loaded after-filters, reflecting both the higher blank levels of certain elements in the polypropylene filter than in the PUF substrate and especially the extremely low mass loading. Though after-filter blanks were similar (Al, Ag, Ce, Mg, Rh, Pt, Sb) or significantly lower (Ba, Ca, Cu, K, Na, Pb, Zn) than for the PUF matrix when equivalent sampled areas are compared, blank concentrations of certain elements in the after-filters were substantially greater than those measured in the PUF substrates. Equivalent area blank concentrations of Cd, Fe, Mn, Ti, V, and Y were over 10 times greater, and concentrations of Cs, Pd, Rb, Sr, Tl, U, and W were 2–6 times greater in the polypropylene filters. It is likely that a modified pre-cleaning protocol, incorporating a dilute HCl rinse, could lower after-filter blank levels, especially considering the properties of the problematic elements. Therefore only a relatively small sub-set of the full suite of metals was judged acceptable for use in this fraction. These elements are: Ca, Al, K, Na, Ba, Cu, Sr, and Pb.
presents a comparison of method detection limits (MDLs) for the 29 elements evaluated in this study. MDLs were calculated as 3 times the propagated analytical uncertainty divided by the sample volume. Analytical uncertainty is the square root of the sum of the variance of multiple substrate section/filter blanks and the square of the uncertainty in the ICP-MS analysis.
TABLE 2 Comparison of method detection limits
The new PUF-ICP-MS method is compared with three other approaches; a 4-hour MOUDI collection, our medium volume PM2.5 approach using pre-cleaned Teflo® substrates coupled with optimized ICP-MS methods, and with dedicated high-quality XRF methods. The MOUDI MDLs reflect 7.2 m3 of air sampled (30 L min−1 for 4 hours), with analytical uncertainties from our optimized ICP-MS methods using pre-cleaned Teflo® substrates. The later two approaches are based on sampling 14 m3 of air over 24 hours, while in the PUF method 180 m3 of air is sampled in 4 hours. The PUF and MOUDI detection limits apply to discrete size-segregated aerosol fractions, while no particle size information (other than the PM2.5 cut) is inherent in the other two methods. For the large majority of elements the PUF-ICP-MS method provides far superior elemental detection capabilities than traditional XRF approaches. The scale of improvement can be 1–2 orders-of-magnitude. A few elements (Ca, Al, Zn, Cu, Cr), where PUF method detection levels are degraded by variability in the PUF matrix blanks, display similar detection limits to the XRF approach, while only two elements (K and Fe) are significantly poorer. PUF method detection limits for many elements are similar to those established for our trace-chemistry optimized medium volume ICP-MS method, and for certain elements (Mn, Sr, Sb, Rb, Ce, Mo, V, Pd, Cd), MDLs are superior. For short interval (4 hours) sampling, the new PUF-ICP-MS method provides significant improvements in elemental detection capability over the MOUDI approach for many of the studied elements (again, as a result of PUF matrix/cleaning blank variability, the MDL's for Ca, Fe, K, Na, Mg, and Ti are degraded with respect to the MOUDI-Teflo® method). Mercury MDLs, particularly those determined from solvent-only cleaned PUF substrates are exceptionally low (0.006 ng m−3). With few exceptions, elemental concentration measured in the PUF fractions from the Phoenix study () were well above our estimated method detection limits.
Table 3 Fine and Coarse concentrations of 28 elements measured in the morning and at midday in Phoenix, AZ on March 25, 2003
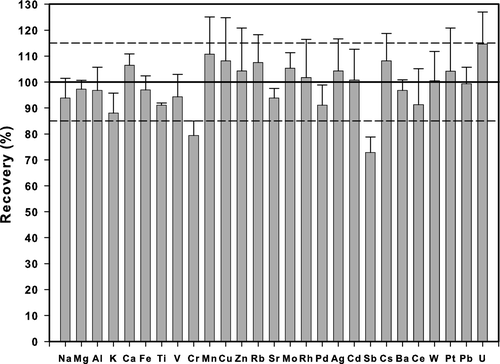
As previously commented the digestion protocol brought into solution and/or converted to CO2 the organic PUF matrix, thus eliminating a factor that could have led to variable analyte responses/recoveries in the ICP-MS analysis. Spike recoveries across all analytes averaged 98.8% (median = 97.6%) with a standard deviation of 8.4%. Individual element average recoveries were all within 10% of target values, with the exception of Ti (87.8%), Mo (85.1%), and Ce (83.8%). Further confirmation of digestion/analysis efficacy is evident at m/z 52, where the molecular species ArC is commonly found—levels of ArC in digested blank PUFs were not significantly different than those in acid method blanks, indicating that the carbon-based PUF matrix was largely removed. Another factor contributing to negligible variation in sample analyte extraction from the plasma is the fact that digestion acid-based ions swamp those contributed by the aerosol PM. Any minor variations in analyte response in this acid matrix are effectively corrected-for by the three internal standards that were used. Analyte recovery from the NIST SRMs, averaged across all 29 reported elements, was 98.5% (median = 97.9%). Individual element average (n = 6 to n = 16 depending upon element) recoveries from the SRMs were all within 10% of certified values, with the exception of K (88.0%), Cr (79.4%), Sb (72.9%), and U (114.6%) (). The variation (standard deviation) in analyte percent recovery, across all elements, averaged 9.4%.
FIG. 2 Mean recoveries of target elements from NIST Standard Reference Materials (Recycled Auto Catalyst (#2556), Urban Dust (#1649a), and San Joaquin Soil (#2709).
Limitations of the method described relate primarily to analyte blank concentrations in the PUF matrix. Blank levels, as they do with all filter-based techniques, will ultimately determine minimum sampling time intervals and the suite of elements quantifiable. As this study demonstrates, despite blank levels typically higher than, for example, Teflon substrates, the PUF method is capable of providing improved MDLs over traditional methods because of the much greater volume of air sampled. A few elements (Sn, Pt) appear to be inherently high in the PUF matrix and cleaning protocols, no matter how sophisticated, may not lower concentrations to acceptable levels. We were not able in this study to address potential variations in PUF blanks across different batches from a given manufacturer or between manufacturers. Clearly, PUF blank levels need to be well characterized to get the most out of the approach. A rigorous comparison of field blanks (including dynamic blanks) with laboratory PUF blanks would be helpful in directing future blank control efforts. However, PUF pre-cleaned with relatively simple techniques is demonstrated to be an acceptable substrate for a large suite of elements. In fact, with some minor changes to the cleaning protocols, it is likely that blank levels of some of the cleaning agent sourced elements could be substantially lowered. The solubilization and quantification techniques described are precise and accurate, but do require careful application of “clean” chemistry. The researcher must be vigilant in the pre-digestion protocols and mass of PUF digested to ensure that over-pressurization of the digestion bombs does not occur. The analytical tools (automated microwave digestion system, and modern ICP-MS) required, while not inexpensive, are being placed in many more laboratories as their advanced capabilities are recognized. This study was not designed to address issues bearing directly on the ChemVol sampler itself, such as stage specific collection efficiencies. The sampler was not designed to provide the detailed size-resolved speciation that a MOUDI sampler is capable of, but the limited PM mass available from any one MOUDI stage severely restricts subsequent characterization. The size-cuts available with the ChemVol are comparable to several commercial personal cascade impactors, therefore comparisons between the two are facilitated.
The overall PUF sampling approach provides a unique tool to cleanly collect large masses of size-resolved PM in short (hourly) time frames—therefore enabling comprehensive chemical characterization on those same intervals. This capability is clearly important in resolving and quantifying PM sources or generation mechanisms that operate on sub-daily cycles. This could include automobile traffic patterns, sources driven by diurnal solar cycles, and in coastal communities, changes in air masses resulting from diurnal shifts in onshore-offshore winds. Importantly, the larger PM masses also make possible the application of advanced techniques such as particle size-resolved stable isotope analysis (e.g., Sr, Pb) or rare-earth ratio methods which show great promise in source reconciliation. While the focus of this study was on developing an approach for quantitative analysis of the total concentrations of a large suite of elements, the PUF method can readily be adapted to selective extractions. In doing so, phase associations, mobility, and bioavailability of elements could be addressed.
Phoenix Demonstration Study
For the demonstration study conducted in Phoenix, Arizona, on March 25, 2003, the sum of the 28 measured elements (with no oxide assumptions) yields coarse (particle diameters between 2.5 and 10 μ m) concentrations of 7.35 ± 0.25 μg m−3 in the morning and 4.49 ± 0.24 μg m−3 during midday and fine (particle diameters less than 2.5 μ m, the sum of the four smallest stages of the sampler) concentrations of 0.91 ± 0.03 μ g m−3 in the morning and 0.56 ± 0.05 μ g m−3 during midday. lists the fine and coarse fraction concentrations of all measured elements in the morning and at midday. For presentation and discussion purposes, the elements are divided into three groups, major (PM10 (sum of fine and coarse) concentration greater than 100 ng m− 3), trace (PM10 concentration between 5 and 100 ng m−3), and ultra-trace (PM10 less than 5 ng m−3).
Concentrations are higher during the morning than at midday, and the trend is not mediated by shifts in just a few elements. In the coarse fraction, all the major elements are significantly higher (p < 0.05) in the morning than midday, and all trace elements and ultra trace elements with measurable concentrations in both morning and midday, are significantly higher (p < 0.05) in the morning than midday except Cd and W. In the fine fraction, about half (9 of 16) of the elements with measurable concentrations in the morning and at midday are significantly higher in the morning, including several major elements, Mg, Fe, and Ti. Of the 7 elements that are not significantly higher in the morning, only Zn and Cd are significantly higher midday than in the morning in the fine fraction. The higher morning concentrations for most elements in both size fractions is expected due to the morning inversion layer (CitationDoran et al. 2003). The few species that exhibit higher midday concentrations, Cd and W in the coarse fraction, and Zn and Cd in the fine fraction are likely due to higher emissions or emissions from different sources reaching the site in the midday. Enrichment factors, used to distinguish anthropogenic elements from soil elements, are calculated for all elements (except Rh and Pt) using Fe as the reference element (submicrometer Al and Si concentrations are below their detection limit) and literature values for the soil composition (CitationTaylor and McLennan 1985). In the midday, the coarse fraction of W and the fine fraction of Zn and Cd have enrichment factors in the 100 s to 1000 s indicating anthropogenic sources for these elements. However the coarse fraction of Cd has a low enrichment factor that does not support an anthropogenic source for the coarse fraction Cd.
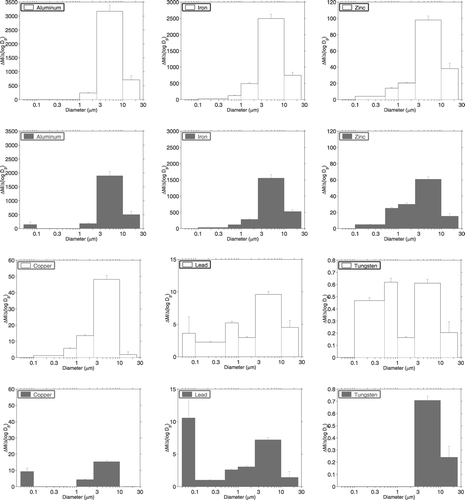
Additional source information can be obtained from size distributions of the elements. shows morning and midday size distributions of two major elements, Al and Fe, three trace elements, Zn, Cu, and Pb and one ultra-trace element, W. Al and Fe as well as Ca, K, Mg, Ti, and Na, exhibit large peaks in the coarse fraction and have low or below detection limit concentrations in the size bins below 1 μ m. These elements are predominately sourced from wind blow soil/dust. Size distributions of some of the trace elements are clearly different than those of the major elements. Peaks between 0.1 and 1.0 μ m are indicative of high temperature sources (CitationOndov and Wexler 1998) and are observed for Mo, Rh, Ag, W, and Pb in the morning and for Cr and Cd during midday. Enrichment factors are 50 to 100,000+ for Cr, Mo, Ag, Cd, W, and Pb in both particle size bins between 0.1 and 1.0 μ m further suggesting their anthropogenic origin. Ultrafine peaks, which are also indicative of anthropogenic sources, are observed for Cu and Rh during midday and for Pb during both sampling periods. Cu, Mo, Ag, W, and Pb have different size distributions during the two time periods and have very large enrichment factors suggesting that different anthropogenic sources impact the site during the morning and midday. Size distributions of some trace elements with known or suspected anthropogenic sources have peaks in the coarse fraction (Cr, Zn, Cu, Sb, W, and Pb), as would be expected for soil-related elements. However, these elements have very large enrichment factors and similar size distributions have been measured for trace elements in tunnel studies (CitationLough et al. 2005) so the large peak in the coarse fraction for these elements is attributed to resuspended road dust.
FIG. 3 Size distributions (ng m−3) of selected major (aluminum and iron), trace (zinc, copper and lead), and ultra-trace (tungsten) elements for morning (white) and midday (black) in Phoenix, Arizona, on March 25, 2003.
CONCLUSIONS
This article described a novel PUF-ICP-MS method for obtaining size segregated ambient aerosol concentrations of a large suite of elements for samples collected over short time periods. This method has the potential to become a powerful tool for identifying sources of elements in the atmosphere and is useful in health effect studies of trace and ultra-trace elements.
The detection capabilities of the 4-hour PUF-ICP-MS method described here were compared to a 4-hour MOUDI approach using optimized ICP-MS. For most elements, the PUF method attained lower MDLs than the MOUDI method. The lower MDLs and higher flowrate of the PUF-ICP-MS method indicates the potential of this method to quantify smaller concentrations of trace elements than the MOUDI-ICP-MS method. Additionally, the PUF-ICP-MS method has lower MDLs than traditional XRF methods for the large majority of elements, suggesting its utility in measuring small elemental concentrations.
The analytical techniques developed for this research enabled the quantification of 28 elements for two size-segregated four-hour samples collected in the morning and during midday on March 25, 2003 in Phoenix, Arizona. The fine and coarse concentrations of most elements were higher in the morning than midday, which has implications for short-term exposure to potentially toxic air pollutants. Measured size distributions of anthropogenic elements such as Cu, W, and Pb show distinct differences between morning and midday indicating that different sources are reaching the sampling site at different times of the day. Twenty-four-hour or 48-hour MOUDI samples are unable to elucidate this short time scale variability in sources and potentially in health effects. Analysis using the PUF-ICP-MS technique for multiple time periods, seasons and locations is needed to accurately identify sources of elements in a city.
We recommend that the future work utilizing the PUF-ICP-MS technique described herein include a characterization of collection efficiency of the sampler stages used.
Acknowledgments
The authors acknowledge Olga Alexandrova for PUF substrate preparation and assistance with sampling, Charity Coury and Xia Su for assistance with sampling and Dustan Helmer and Joel Overdier for their detailed and dedicated laboratory assistance.
Notes
a < Limit of Detection.
b One (of six) values rejected as an outlier in both Full and Solvent cleaning sets.
a All MDLs are expressed as 3x the propagated uncertainty.
b MDLs reflect error components from PUF substrate variation, ICP-MS analysis and sampling 180 m3.
c MDLs calculated using propagated uncertainty from UW-Madison ICP-MS method4 (pre-cleaned 47 mm Teflon substrates) and sampling 7.2 m3 (30 L min− 1 for 4 hours).
d UW-Madison ICP-MS method using pre-cleaned 47mm Teflo filter substrate, sampling 14 m3. (CitationLough et al. 2005).
e XRF limits from (CitationChow 1995); 14 m3 sample.
a f NA = Not analyzed.
g Mercury analysis by CVAF. The two 4-hour PUF MDL values given reflect “Full” cleaning and “Solvent-only” cleaning respectively.
a bd = value below method detection limit or less than measurement uncertainty.
REFERENCES
- Carlton , A. G. , Turpin , B. J. , Johnson , W. , Buckley , B. T. , Simcik , M. , Eisenreich , S. J. and Porcja , R. J. 1999 . Microanalysis Methods for Characterization of Personal Aerosol Exposures . Aerosol Sci. Technol. , 31 : 66 – 80 .
- Chow , J. C. 1995 . Measurement Methods to Determine Compliance with Ambient Air–Quality Standards for Suspended Particles . J. Air & Waste Manage. Assoc. , 45 : 320 – 382 .
- Demokritou , P. , Kavouras , I. G. , Ferguson , S. T. and Koutrakis , P. 2002 . Development of a High Volume Cascade Impactor for Toxicological and Chemical Characterization Studies . Aerosol Sci. Technol. , 36 : 925 – 933 .
- Dillner , A. M. , Schauer , J. J. , Christensen , W. F. and Cass , G. R. 2005 . A Quantitative Method for Clustering Size Distributions of Elements . Atmos. Environ. , 39 : 1525 – 1537 .
- Doran , J. C. , Berkowitz , C. M. , Coulter , R. L. , Shaw , W. J. and Spicer , C. W. 2003 . The 2001 Phoenix Sunrise Experiment: Vertical Mixing and Chemistry During the Morning Transition in Phoenix . Atmos. Environ. , 37 : 2365 – 2377 .
- Graney , J. R. , Landis , M. S. and Norris , G. A. 2004 . Concentrations and Solubility of Metals from Indoor and Personal Exposure PM2.5 Samples . Atmos. Environ. , 38 : 237 – 247 .
- Lingard , J. J. N. , Tomlin , A. S. , Clarke , A. G. , Healey , K. , Hay , A. W. M. , Wild , C. P. and Routledge , M. N. 2005 . A Study of Trace Metal Concentration of Urban Airborne Particulate Matter and Its Role in Free Radical Activity as Measured by Plasmid Strand Break Assay . Atmos. Environ. , 39 : 2377 – 2384 .
- Lough , G. C. , Schauer , J. J. , Park , J. S. , Shafer , M. M. , Deminter , J. T. and Weinstein , J. P. 2005 . Emissions of Metals Associated with Motor Vehicle Roadways . Environ. Sci. Technol. , 39 : 826 – 836 .
- Oberdorster , G. 2001 . Pulmonary Effects of Inhaled Ultrafine Particles . Int. Arch. Occup. Environ. Health. , 74 : 1 – 8 .
- Ondov , J. M. and Wexler , A. S. 1998 . Where Do Particulate Toxins Reside? An Improved Paradigm for the Structure and Dynamics of the Urban Mid–Atlantic Aerosol . Environ. Sci. Technol. , 32 : 2547 – 2555 .
- Peters , A. , Dockery , D. W. , Muller , J. E. , Murray , A. and Mittleman , M. D. 2001 . Increased Particulate Air Pollution and the Triggering of Myocardial Infarction . Circulation. , 103 : 2810 – 2815 .
- Pyle , S. M. , Nocerino , J. M. , Deming , S. N. , Palasota , J. A. , Palasota , J. M. , Miller , E. L. , Hillman , D. C. , Kuharic , C. A. , Cole , W. H. , Fitzpatrick , P. M. , Watson , M. A. and Nichols , K. D. 1996 . Comparison of Aas, Icp–Aes, Psa, and Xrf in Determining Lead and Cadmium in Soil . Environ. Sci. Technol. , 30 : 204 – 213 .
- Riley , M. R. , Boesewetter , D. E. , Kim , A. M. and Sirvent , F. P. 2003 . Effects of Metals Cu, Fe, Ni, V, and Zn on Rat Lung Epithelial Cells . Toxicology. , 190 : 171 – 184 .
- Smith , F. E. and Arsenault , E. A. 1996 . Microwave–Assisted Sample Preparation in Analytical Chemistry . Talanta. , 43 : 1207 – 1268 .
- Swami , K. , Judd , C. D. , Orsini , J. , Yang , K. X. and Husain , L. 2001 . Microwave Assisted Digestion of Atmospheric Aerosol Samples Followed by Inductively Coupled Plasma Mass Spectrometry Determination of Trace Elements . Fresenius Journal of Analytical Chemistry. , 369 : 63 – 70 .
- Taylor , S. R. and McLennan , S. M. 1985 . The Continental crust: Its composition and evolution: An examination of the geochemical record preserved in sedimentary rocks , 312 Oxford : Blackwell Scientific .
- U.S. EPA . 1999 . “ Method IO-3.5: Determination of Metals in Ambient Particulate Matter Using Inductively Coupled Plasma/Mass Spectrometry (ICP/MS) ” . In Compendium of Methods for Chemical Species Analysis of Filter Collected SPM , Cincinnati , OH : Center for Environmental Research and Development, ORD .
- Wang , C. F. , Chang , E. E. , Chiang , P. C. and Aras , N. K. 1995a . Analytical Procedures on Multielement Determinations of Airborne Particles for Receptor Model Use . Analyst. , 120 : 2521 – 2527 .
- Wang , C. F. , Chen , W. H. , Yang , M. H. and Chiang , P. C. 1995b . Microwave Decomposition for Airborne Particulate Matter for the Determination of Trace–Elements by Inductively–Coupled Plasma–Mass Spectrometry . Analyst. , 120 : 1681 – 1686 .
- Yang , K. X. , Swami , K. and Husain , L. 2002 . Determination of Trace Metals in Atmospheric Aerosols with a Heavy Matrix of Cellulose by Microwave Digestion–Inductively Coupled Plasma Mass Spectroscopy . Spectrochimica Acta Part B–Atomic Spectroscopy. , 57 : 73 – 84 .