Abstract
Concentrations of fine carbonaceous aerosols (PM2.5), including elemental carbon (EC), organic carbon (OC), and water-soluble organic carbon (WSOC) were measured from filters collected every 6th day for one year at three urban/industrial sites in the Midwestern United States: Cincinnati, Cleveland, and Mingo Junction, Ohio. The water-soluble fraction of fine particulate OC varied considerably from site to site and monthly averages were between 39–85% at Cincinnati, 35–68% at Cleveland, and 32–65% at Mingo Junction. Average monthly concentrations of WSOC were compared with measurements of organic source tracers, including levoglucosan, to better understand the spatial and temporal distribution and sources of WSOC. Two methods of predicting the non-biomass burning portion of WSOC (WSOCNB) were compared including estimation of secondary organic carbon (SOC) by the EC-tracer Method and comparison of the unapportioned OC from a chemical mass balance (CMB) source apportionment analysis. Poor correlations between SOC estimated by the EC-tracer method and WSOCNB suggested that the use of the EC-tracer method to estimate SOC may be significantly flawed with respect to low time-resolved measurements, such as one-in-six day measurements. Good correlations between CMB unapportioned OC and WSOCNB at all three sites (R2 = 0.68 to 0.91) suggested that direct measurements of levoglucosan and WSOC could provide a reasonable estimate of secondary organic carbon concentrations in some locations. However, application of this method to daily measurements made in Detroit, MI during July of 2007 and January/February of 2008 demonstrated that, on some individual days near large point sources, non-biomass burning sources of WSOC were important contributors to WSOC concentrations.
1. INTRODUCTION
Given that organic aerosols can comprise from 10 to 70% of the total fine aerosol mass in many locations (CitationAndrews et al. 2000), understanding the sources of organic carbon (OC) has become an important focus for investigators. While source apportionment methods based on the quantification of molecular markers have been effective in understanding the sources of condensed-phase OC that is directly emitted into the atmosphere (e.g., CitationHildemann et al. 1991; CitationSchauer et al. 1996; CitationZheng et al. 2002), known as primary OC, these methods have not typically been used to directly resolve the fraction of OC that is derived from the photo-oxidation and subsequent condensation of anthropogenic and biogenic gas-phase emissions, known as secondary OC. Although several potential tracers have been identified (CitationSchauer et al. 2002; CitationSheesley et al. 2004; CitationKleindienst et al. 2007) and used in recent studies to apportion the contribution of secondary organic carbon (SOC) to fine atmospheric particulate matter (CitationShrivastava et al. 2007; CitationLewandowski et al. 2008; CitationStone et al. 2009), the use of these tracers has not been fully validated and is not generally accepted as a robust source apportionment tool. Quantifying the contribution of secondary organic aerosols (SOA) to OC concentrations is important to understanding whether source-apportionment models account for all of the significant primary sources of OC at a given receptor site and is a key component in assessing air pollution control strategies.
Previous work has shown that SOA is primarily composed of oxygenated compounds that are highly to moderately water-soluble (CitationKanakidou et al. 2005 and references therein). Accordingly, measurements of the water-soluble organic carbon (WSOC) have been used in the past to estimate the contribution of secondary organic aerosols and OC from biomass smoke, a large fraction of which is also highly water soluble (CitationSannigrahi et al. 2006), to PM2.5 organic carbon concentrations (CitationDecesari et al. 2001; CitationSzidat et al. 2004; CitationDocherty et al. 2008; CitationStone et al. 2008). However, these studies have been primarily focused on regional haze events and short-term intensive studies, and there is a need to develop a seasonally based understanding of the sources of WSOC in order to evaluate its potential use in estimating SOC.
In addition to its potential as a general tracer for biomass burning emissions and SOC, WSOC may play an important role in the heterogeneous chemistry that occurs in fog and cloud water (CitationFuzzi et al. 2002; CitationCappiello et al. 2003), which has implications for local and regional air quality. WSOC can potentially alter the hygroscopic properties of ambient aerosols making them more effective cloud condensation nuclei (CitationSaxena et al. 1995) and thus may have an impact on global climate. In addition, the water-soluble components of particulate matter are more easily absorbed into lung fluids potentially enhancing their ability to produce adverse human health effects. For these reasons, understanding the sources and spatial/temporal patterns of WSOC are important to both climate and toxicity studies.
In order to better understand the contributions of SOC and biomass burning to WSOC, samples collected at three sites located in the Midwestern United States: Cincinnati, Cleveland, and Mingo Junction, OH, during a year-long study were analyzed for fine particulate EC, OC, WSOC, and primary organic source tracers. Estimates of secondary organic carbon were derived using the EC-tracer Method and using the unapportioned OC mass from a chemical mass balance (CMB) source apportionment analysis. Results obtained from these methods indicate that on a monthly averaged basis, measurements of levoglucosan and WSOC can provide reasonable estimates of SOC at some locations. Application of these methods to samples collected in Detroit, MI, however, indicate that emissions from some industrial sources can also make significant contributions to fine particulate WSOC concentrations at locations near these sources.
2. EXPERIMENTAL
Concentrations of fine carbonaceous aerosols (PM2.5), including elemental carbon (EC), organic carbon (OC), and water-soluble organic carbon (WSOC) were measured for one year at three urban/industrial sites in the Midwestern United States: Cincinnati, Cleveland, and Mingo Junction, Ohio. Sites were selected in order to obtain a regional perspective on the variability and sources of carbonaceous aerosols. The Cincinnati site was located in a mixed business/residential district with few nearby industrial sources; the Cleveland site was located near busy surface streets, a highway interchange, and near several heavy industrial sources situated along the Cuyahoga River; and the Mingo Junction site was located in a rural/suburban neighborhood within the Ohio River Valley, proximate to numerous industrial sources. At the time of this study, all three sites were in non-attainment with regard to the United States Environmental Protection Agency's national ambient air quality standards (NAAQS) for fine particulate matter (PM2.5).
Twenty-four hour integrated PM2.5 samples were also collected on a daily basis during two, one-month intensive sampling periods at three sites located in the Detroit, MI metropolitan area. These sites were located at Newberry Elementary on the near-west side of Detroit, at Salina Middle School in Dearborn, MI and at a monitoring site located in Allen Park, MI. All three sites are adjacent to heavy and light industry, including steel production and automobile manufacturing facilities, and are located along the I-75/ I-94 corridor that parallels the Detroit River.
2.1. Sample Collection and Analysis
Twenty-four hour midnight-to-midnight CST samples of fine particulate matter (PM2.5) were collected on the EPA 1-in-6 day schedule from June of 2007 through May of 2008 at three sites in the Midwestern United States: Cincinnati, Cleveland, and Mingo Junction, Ohio. Daily PM2.5 samples were also collected at three sites in Detroit, Michigan during July 2007 and January/ February 2008 as a part of an intensive sampling effort. Aerosol samples were collected on pre-baked 90 mm quartz-fiber filters (Pall Gellman, Ann Arbor, MI) at each site by a URG-3000B particulate sampler (URG, Chapel Hill, NC). Filters were stored in sealed Petri dishes lined with baked aluminum foil and kept in a freezer at or below −5°C before and after sampling. The URG samplers were operated at 92 L min–1 and were each fitted with a PM2.5 cyclone.
Samples and field blanks were analyzed for organic carbon and elemental carbon content using a Thermal Optical Analyzer (Sunset Laboratory, Forest Grove OR) using the ACE-Asia method (CitationSchauer et al. 2003), and monthly composites of 1-in-6 day samples were analyzed for non-polar organic markers and levoglucosan by gas-chromatography/mass spectrometry (GCMS) following the methods outlined by CitationStone et al. (2008). Filters were also analyzed for water-soluble organic carbon (WSOC) content by extracting a 1.5 cm2 punch from each filter. Filter punches were placed in acid-washed centrifuge tubes along with purified water and agitated on a shaker table for two hours at room temperature. After agitation, samples were then filtered through acid-washed, 0.2 μm polypropylene syringe filter and analyzed for organic carbon using a Shimadzu TOC-5000A total organic carbon analyzer, which utilizes a high temperature combustion technique (CitationWangersky 1993).
2.2. Chemical Mass Balance (CMB) Source Apportionment Model
Source apportionment calculations were accomplished using a chemical mass balance (CMB) model. A detailed description of this model and results are available in CitationRutter et al. (2008) and will only be briefly discussed here. Primary source contributions to PM2.5 organic carbon were determined using a molecular marker approach in which contributions were determined from the effective-variance least squares linear regression of the product of the source-contributions and concentrations of a set of organic molecular markers (CitationWatson et al. 1984). Calculations of source contributions were accomplished using a software package (EPA-CMB v. 8.2) available from the United States Environmental Protection Agency. Specific markers used in this analysis were, EC, C28-C34 n-alkanes, levoglucosan, 17α (H)-22,29,30-trisnorhopane, 17β (H)-21α (H)-30-norhopane, 17α (H)-21β (H)-hopane, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(e)pyrene, indeno(1,2,3-cd)pyrene, benzo(ghi)perylene, and picene.
It should be noted that some uncertainty in the apportionment of OC to primary sources exists due to the sensitivity of CMB to the type and composition of source profiles used (CitationSubramanian et al. 2006; CitationLee and Russell 2007; CitationLough and Schauer 2007; CitationRutter et al. 2008). Relevant to the work presented here, CitationSheesley et al. (2007) demonstrated that although the apportionment of OC to biomass burning by CMB is sensitive to the choice of source profile (the reported standard deviation across 5 source profiles was a little greater than 30%), the uncertainty due to the choice of profile is similar to the underlying analytical uncertainty of the levoglucosan measurements, providing confidence in the apportionment of OC to biomass burning. This same study concluded that the choice of biomass burning profile can affect the attribution of OC to other sources, including the amount of OC unapportioned by CMB; however, CitationSheesley et al. (2007) also concluded that the impact was minimal when using a geographically relevant profile.
Although recent chamber studies have indicated the potential for the oxidation/degradation of some of the tracers used in the current study, there is, as of yet, no compelling evidence that under atmospherically relevant conditions and concentrations aging of molecular markers occurs that would cause significant underestimation of primary sources by CMB.
2.3. EC Tracer Model
Secondary organic carbon concentrations were predicted by using EC as a tracer for primary combustion sources. This technique has been used to distinguish primary OC from secondary OC in numerous studies (CitationTurpin and Huntzicker 1991; CitationTurpin et al. 1991; CitationTurpin and Huntzicker 1995; CitationCastro et al. 1999; CitationStrader et al. 1999; CitationLim and Turpin 2002b; CitationRussell and Allen 2004; CitationYu et al. 2004) and generally involves the assumption that the primary emissions of OC and EC have common sources, and that at a given site, the ratio of OC/EC emitted from primary sources (hereafter referred to as [OC/EC]prim) can be determined from periods in which conditions are highly unfavorable for the production of SOC. Assuming that OC is the result of primary combustion emissions (OCcomb), primary non-combustion emissions (OCnon - comb), and secondary organic carbon formation, the EC-tracer Method uses Equation (Equation1) to calculate SOC at a given receptor.
Numerous methods of determining the value of [OC/EC]prim and OCnon - comb have been used in previous studies, including simple slope estimations. The most common approach is a linear regression of a subset of data in which [OC/EC]prim is determined as the slope and OCnon - comb as the y-intercept (CitationSaylor et al. 2006). For this work, regression based on the work of CitationDeming (1943), which assumes, unlike conventional linear regression, that error is present in both variables being regressed, was used to determine the values of [OC/EC]prim and OCnon - comb from the lowest 5% of OC/EC values observed at each site (1-in-6 day samples, n = 4 for each site). Values of [OC/EC]prim calculated for this analysis (Cincinnati = 5.74, Cleveland = 3.83, and Mingo Junction = 4.44) were generally higher than what has been previously reported, which range from ∼1.2 to 2 (CitationCastro et al. 1999; CitationCabada et al. 2004; CitationPolidori et al. 2007). Calculated values for OCnon - comb were 0.31 μg m–3 at Cincinnati, 0.15 μg m–3 at Cleveland, and 0.26 μg m–3 at Mingo Junction.
3. RESULTS
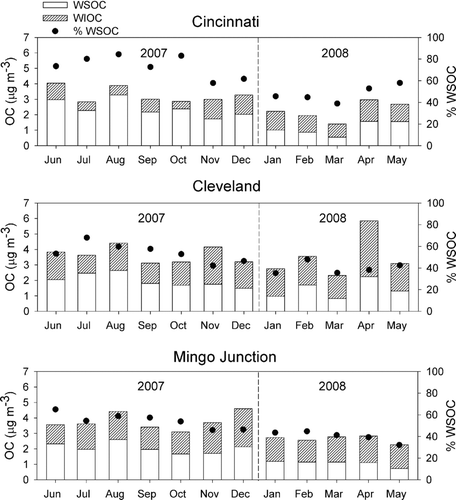
A comparison of the seasonal variations in PM2.5 WSOC and water insoluble organic carbon (WIOC = OC–WSOC), shown in , reveals that WSOC was higher on a monthly averaged basis at all three sites during the summer and early fall, both in terms of absolute concentrations and percentage of OC, which is consistent with the photochemical production of SOC. Spatially, WSOC concentrations showed significant site-to-site variability indicative of the influence of local primary sources of WSOC, such as biomass burning, or differences in the emissions of precursors to secondary organic aerosol formation. Overall, the average concentration of water-insoluble OC observed in Cincinnati was significantly lower (1.01 ± 0.08 μg m–3) than what was observed in Cleveland (1.81 ± 0.14 μg m–3) and Mingo Junction (1.66 ± 0.13 μg m–3), possibly due to greater industrial activity in and around the latter two cities.
FIG. 1 Monthly average 1-in-6 day PM2.5 water soluble organic carbon (WSOC) and water insoluble organic carbon (WIOC).
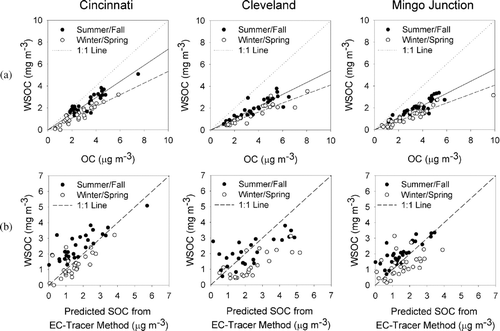
As shown in , the relationship between WSOC and OC appeared to have distinct seasonal differences such that the WSOC/OC ratio was distinctly higher during the summer/fall months (June–November) than during the winter/spring months (December–May). In order to better understand the nature of WSOC observed at these sites, an estimate of SOC was determined using the EC-tracer Method, as described previously, and compared with the observed WSOC concentrations. The EC-racer analysis, illustrated in , resulted in a poor correlation between predicted SOC and WSOC. This suggested that either there were significant contributions to WSOC from primary sources, even during the summer months, or that the one-in-six day sampling period did not provide an opportunity to obtain a reasonable estimate of [OC/EC]prim.
FIG. 2 (a) Seasonal comparison of PM2.5 OC and WSOC. Solid lines represent the regression of the 1-in-6 day summer/fall observations and dashed lines represent the 1-in-6 day spring observations. (b) Seasonal comparison of 1-in-6 day observations of PM2.5 WSOC with SOC as predicted by the EC tracer method. Regression statistics for these plots are shown in Table S2 in the supplemental materials accompanying this article.
As organic carbon emissions from biomass burning have also been found be to highly water-soluble, estimates of WSOC emissions from biomass burning were calculated from OC apportioned to biomass burning by a molecular marker—CMB analysis in order to try and reconcile the discrepancies observed in the EC-tracer model. Estimations of the contribution of biomass burning to OC ranged from 1.8 to 58.8% at Cincinnati, 4.8 to 61.2% at Cleveland, and 5.4 to 62.6% at Mingo Junction. Previous work by CitationSannigrahi et al. (2006), in which filter-based aerosol samples were collected next to prescribed burns, concluded that the majority of OC emitted from biomass burning is water soluble. Based on their work, which determined that WSOC comprised approximately 71% of OC emitted by biomass burning, the amount of biomass-burning WSOC (hereafter referred to as WSOCBB) was determined as 71% of the OC apportioned to biomass burning by the CMB model. This amount was then subtracted from the measured WSOC to provide an estimate of non-biomass burning WSOC (hereafter referred to as WSOCNB), assumed to be primarily composed of secondary organic carbon (CitationLim and Turpin 2002a; CitationWeber et al. 2007).
WSOCNB was then compared with monthly averaged SOC as predicted by the EC-tracer analysis and with unapportioned organic aerosol (sometimes referred to as CMB “Other”). The results of the comparison between WSOCNB and predicted SOC from the EC-tracer Method () were generally poor (Cincinnati R2 = 0.36, Cleveland R2 = 0.04, Mingo Junction, R2 = 0.14) with good correlation seen only during the summer/fall at the Mingo Junction site. In general, better correlations between SOC predicted by the EC tracer method and WSOCNB were observed during the summer/fall period than in the winter/spring, similar to what has been observed previously between CMB unapportioned OC and SOC predicted by the EC tracer method (CitationSubramanian et al. 2007). The results of this comparison reveal that there are significant limitations to the use of the EC-tracer method. Specifically, estimation of [OC/EC]prim from low time-resolved data sets appears to be problematic due to temporal variations in combustion emissions and the effect of sampling frequency, which may also affect the value of OCnon - comb (Gray et al. 1986; Cabada et al. 2002; CitationCabada et al. 2004). In the case of this analysis, it is likely that the one-in-six day sampling frequency was not adequate for determining [OC/EC]prim, as this ratio may change on time scales finer than one-in-six-day measurements can adequately represent. In addition, using a single value for [OC/EC]prim may ignore significant seasonal variations in primary aerosol composition, which may produce large differences in [OC/EC]prim. In addition, finding a representative [OC/EC]prim may be especially difficult during summer months when OC may be dominated by SOC.
FIG. 3 Seasonal comparison of measured PM2.5 non-biomass burning WSOC (WSOCNB) and (a) SOC as predicted by the EC-tracer method and (b) SOC predicted by CMB “other.”
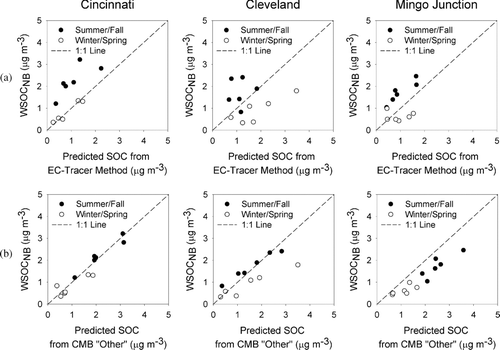
Values of [OC/EC]prim were also calculated from the results of the CMB analysis using Equation (Equation2). The average value of [OC/EC]prim determined for each site was within the range of the values determined from the observed OC and EC concentrations (Cincinnati = 4.5, Cleveland = 4.6, and Mingo Junction = 3.6). However, the ratios determined from the CMB model showed significant seasonal and month-to-month variability (refer to Table S1 in the supplemental material) suggesting that the use of a single [OC/EC]prim value across a year-long study period may lead to significant errors in the estimation of SOC, which may help explain the poor correlation between SOC estimated by the EC-tracer method and WSOCNB.
As shown in , comparisons between WSOCNB and CMB “other” were significantly better than what was observed with the EC-tracer Method. Strong correlations were seen across all three sites (Cincinnati R2 = 0.91, Cleveland R2 = 0.68, Mingo Junction, R2 = 0.89), with some scatter observed at the Cleveland site. The relationship between CMB “other” and WSOCNB at Mingo Junction was strong, although it deviated significantly from the 1:1 line. This may reflect the influence of uncharacterized local sources of OC or a difference in the WSOC content of local biomass-burning emissions. In general, these results suggest that WSOC observed at the Cincinnati, Cleveland, and Mingo Junction sites was principally composed of water-soluble organic carbon derived from secondary organic carbon and biomass-burning smoke. This analysis further suggests that the contributions to WSOC from these two sources may be readily estimated from direct measurements of levoglucosan, a well-established tracer for biomass-burning emissions (CitationSimoneit 2002), and WSOC.
Estimates of the contributions of biomass burning and SOA to WSOC concentrations using measurements of levoglucosan and WSOC for sites in Cincinnati, Cleveland, and Mingo Junction are shown in . These results demonstrate that the contribution of biomass burning to WSOC had a seasonal pattern at these sites during which maximum contributions were observed during the winter months. Specifically, between 55 and 60% of WSOC was attributed to biomass-burning sources during the months of December and January at all three sites, which represent roughly 25% of the observed total OC. The observed concentrations and spatial heterogeneity of WSOCBB are consistent with the impact of biomass smoke from winter-time residential wood burning.
FIG. 4 Monthly estimates of the contribution of biomass burning to PM2.5 WSOC (WSOCBB).
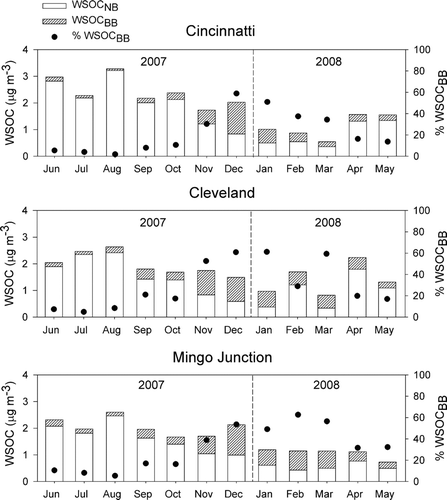
The contribution of SOA to WSOC was highest during the summer months when the greatest photochemical activity was expected. During the months of June, July, and August, 90% or greater of the observed WSOC was attributed to secondary aerosols, which accounted for greater than three quarters of total OC at the Cincinnati site and roughly half of the observed total OC at the Cleveland and Mingo Junction sites. These results clearly demonstrate that SOC is the single greatest contributor to OC mass during the summer at all three locations.
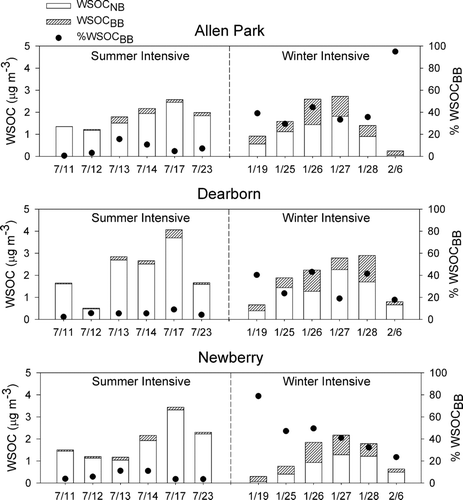
Estimates of the contributions of biomass smoke and SOA to WSOC concentrations were also made using daily measurements of carbonaceous aerosols collected during July of 2007 and January/February of 2008 at three sites in the Detroit, MI metropolitan area. Although WSOC measurements were made on each daily sample, only a limited number of samples were examined for organic molecular markers (e.g., levoglucosan). As shown in , the greatest contributions to WSOC from biomass burning were again observed in the winter samples, although there were clearly significant contributions to WSOC from SOA on many of the winter days examined in this study. The daily contribution of biomass smoke to WSOC ranged from as much as 95% to as little as 20% of WSOC. The general pattern in SOA contributions was similar at two of the sites, Allen Park and Newberry, during both the summer and winter days; however, several days at the Dearborn site showed significantly higher non-biomass burning contributions suggesting the presence of a local source of WSOC. These observations were consistent with evidence that WSOC concentrations were generally higher at the Dearborn site than at either the Allen Park or Newberry Sites, particularly during the January/February study period (see Supplementary ). By assuming that the average of WSOCNB at Allen Park and Newberry represented the average urban levels of SOC, it was calculated that the local source(s) of water-soluble organic carbon impacting the Dearborn site may have contributed between 0.0 and 1.4 μg m–3 to the concentration of WSOC on the observed days.
FIG. 5 Estimates of the contribution of biomass burning to PM2.5 WSOC (WSOCBB) during selected days at three sites in Detroit, MI.
4. DISCUSSION
On a monthly averaged basis, measurements of levoglucosan and WSOC provide a relatively simple method of estimating the contributions of biomass burning and SOA to WSOC in fine atmospheric particulate matter at some locations. The use of the WSOCNB as a proxy for secondary organic carbon, however, is predicated on the assumption that the two most significant sources of WSOC to fine atmospheric particulate matter are SOA and biomass smoke. While this may be true in some locations on a monthly, seasonal, or annual basis, this work indicates that emissions from primary, non-biomass burning sources of water-soluble organic carbon can contribute significantly to the day-to-day variability and concentration of organic carbon observed in urban/industrialized locations. The results of this study indicate that primary sources of WSOC, including the potential aging of primary emissions, need to be better characterized in order to better understand the temporal and spatial variability of WSOC. In addition, these results strongly suggest that future carbonaceous aerosol source apportionment efforts would greatly benefit from the integration of WSOC measurements.
Supplemental Material
Download Zip (153.6 KB)Acknowledgments
[Supplementary materials are available for this article. Go to the publisher's online edition of Aerosol Science and Technology to view the free supplementary files.]
The authors wish to acknowledge the extraordinary co-operation of Dan Ling and his staff at the Michigan Department of Environmental Quality and the assistance of the Dearborn Public Schools. We also acknowledge the assistance of the Cleveland Department of Public Health's Division of Air Quality, the Ohio Environmental Protection Agency, and the Hamilton County Department of Environmental Services. We would also like to acknowledge and thank Martin Shafer, Brian Majestic, and Mike Olson for assistance with WSOC/TOC analysis and with sample collection during the winter intensive study in Detroit. Funding for this study was provided by the Lake Michigan Air Director's Consortium (LADCO).
REFERENCES
- Andrews E.Saxena P.Musarra S.Hildemann L. M.Koutrakis P.McMurry P. H.Olmez I.White W. H.Concentration and Composition of Atmospheric Aerosols from the 1995 SEAVS Experiment and a Review of the Closure Between Chemical and Gravimetric MeasurementsJ. the Air & Waste Manage. Assoc.200050648–664
- Cabada J. C.Pandis S. N.Subramanian R.Robinson A. L.Polidori A.Turpin B.Estimating the Secondary Organic Aerosol Contribution to PM2.5 Using the EC Tracer MethodAerosol Sci. Technol.200438140–155
- Cappiello A.De Simoni E.Fiorucci C.Mangani F.Palma P.Trufelli H.Decesari S.Facchini M. C.Fuzzi S.Molecular Characterization of the Water-Soluble Organic Compounds in Fogwater by ESIMS/MSEnviron. Sci. & Technol.2003371229–1240
- Castro L. M.Pio C. A.Harrison R. M.Smith D. J. T.Carbonaceous Aerosol in Urban and Rural European Atmospheres: Estimation of Secondary Organic Carbon ConcentrationsPergamon-Elsevier Science Ltd.19992771–2781
- Decesari S.Facchini M. C.Matta E.Lettini F.Mircea M.Fuzzi S.Tagliavini E.Putaud J. P.Chemical Features and Seasonal Variation of Fine Aerosol Water-Soluble Organic Compounds in the Po Valley, ItalyAtmos. Environ.2001353691–3699
- Deming W. E.Statistical Adjustment of DataWileyLondon1943
- Docherty K. S.Stone E. A.Ulbrich I. M.DeCarlo P. F.Snyder D. C.Schauer J. J.Peltier R. E.Weber R. J.Murphy S. N.Seinfeld J. H.Grover B. D.Eatough D. J.Jiimenez J. L.Apportionment of Primary and Secondary Organic Aerosols in Southern California during the 2005 Study of Organic Aerosols in Riverside (SOAR-1)Environ. Sci. & Technol.2008427655–7662
- Fuzzi S.Facchini M. C.Decesari S.Matta E.Mircea M.Soluble Organic Compounds in Fog and Cloud Droplets: What have we Learned Over the Past Few Years?Atmos. Res.20026489–98
- Hildemann L. M.Markowski G. R.Cass G. R.Chemical-Composition of Emissions from Urban Sources of Fine Organic AerosolEnviron. Sci. & Technol.199125744–759
- Kanakidou M.Seinfeld J. H.Pandis S. N.Barnes I.Dentener F. J.Facchini M. C.Van Dingenen R.Ervens B.Nenes A.Nielsen C. J.Swietlicki E.Putaud J. P.Balkanski Y.Fuzzi S.Horth J.Moortgat G. K.Winterhalter R.Myhre C. E. L.Tsigaridis K.Vignati E.Stephanou E. G.Wilson J.Organic Aerosol and Global Climate Modelling: A ReviewAtmos. Chem. Phys.200551053–1123
- Kleindienst T. E.Jaoui M.Lewandowski M.Offenberg J. H.Lewis C. W.Bhave P. V.Edney E. O.Estimates of the Contributions of Biogenic and Anthropogenic Hydrocarbons to Secondary Organic Aerosol at a Southeastern US LocationAtmos. Environ.2007418288–8300
- Lee S.Russell A. G.Estimating Uncertainties and Uncertainty Contributors of CMB PM2.5 Source Apportionment ResultsAtmos. Environ.2007419616–9624
- Lewandowski M.Jaoui M.Offenberg J. H.Kleindienst T. E.Edney E. O.Sheesley R. J.Schauer J. J.Primary and Secondary Contributions to Ambient PM in the Midwestern United StatesEnviron. Sci. & Technol.2008423303–3309
- Lim H. J.Turpin B. J.Origins of Primary and Secondary Organic Aerosol in Atlanta: Results' of Time-Resolved Measurements During the Atlanta Supersite ExperimentEnviron. Sci. & Technol.2002a364489–4496
- Lim H. J.Turpin B. J.Origins of Primary and Secondary Organic Aerosol in Atlanta: Results of Time-Resolved Measurements During the Atlanta Supersite ExperimentEnviron. Sci. & Technol.2002b364489–4496
- Lough G. C.Schauer J. J.Sensitivity of Source Apportionment of Urban Particulate Matter to Uncertainty in Motor Vehicle Emissions ProfilesJ. Air & Waste Manage. Assoc.2007571200–1213
- Polidori A.Arhami M.Sioutas C.Delfino R. J.Allen R.Indoor/Outdoor Relationships, Trends, and Carbonaceous Content of Fine Particulate Matter in Retirement Homes of the Los Angeles BasinJ. Air & Waste Manage. Assoc.200757366–379
- Russell M.Allen D. T.Seasonal and Spatial Trends in Primary and Secondary Organic Carbon Concentrations in Southeast TexasAtmos. Environ.2004383225–3239
- Rutter A. P.Snyder D. C.Schauer J. J.Deminter J. T.Shelton B.The Impact of Coal Combustion on the Source Apportionment of Organic Carbon in Atmospheric Particulate MatterEnviron. Sci. & Technol.2008 submitted
- Sannigrahi P.Sullivan A. P.Weber R. J.Ingall E. D.Characterization of Water-Soluble Organic Carbon in Urban Atmospheric Aerosols Using Solid-State C-13 NMR SpectroscopyEnviron. Sci. & Technol.200640666–672
- Saxena P.Hildemann L. M.Mcmurry P. H.Seinfeld J. H.Organics Alter Hygroscopic Behavior of Atmospheric ParticlesJ. Geophys. Res.–Atmospheres199510018755–18770
- Saylor R. D.Edgerton E. S.Hartsell B. E.Linear Regression Techniques for Use in the EC Tracer Method of Secondary Organic Aerosol EstimationAtmos. Environ.2006407546–7556
- Schauer J. J.Fraser M. P.Cass G. R.Simoneit B. R. T.Source Reconciliation of Atmospheric Gas-Phase and Particle-Phase Pollutants During a Severe Photochemical Smog EpisodeEnviron. Sci. & Technol.2002363806–3814
- Schauer J. J.Mader B. T.Deminter J. T.Heidemann G.Bae M. S.Seinfeld J. H.Flagan R. C.Cary R. A.Smith D.Huebert B. J.Bertram T.Howell S.Kline J. T.Quinn P.Bates T.Turpin B.Lim H. J.Yu J. Z.Yang H.Keywood M. D.ACE-Asia Intercomparison of a Thermal-Optical Method for the Determination of Particle-Phase Organic and Elemental CarbonEnviron. Sci. & Technol.200337993–1001
- Schauer J. J.Rogge W. F.Hildemann L. M.Mazurek M. A.Cass G. R.Source Apportionment of Airborne Particulate Matter Using Organic Compounds as TracersAtmos. Environ.1996303837–3855
- Sheesley R. J.Schauer J. J.Bean E.Kenski D.Trends in Secondary Organic Aerosol at a Remote Site in Michigan's Upper PeninsulaEnviron. Sci. & Technol.2004386491–6500
- Sheesley R. J.Schauer J. J.Zheng M.Wang B.Sensitivity of Molecular Marker–Based CMB Models to Biomass Burning Source ProfilesAtmos. Environ.2007419050–9063
- Shrivastava M. K.Subramanian R.Rogge W. F.Robinson A. L.Sources of Organic Aerosol: Positive Matrix Factorization of Molecular Marker Data and Comparison of Results from Different Source Apportionment ModelsAtmos. Environ.2007419353–9369
- Simoneit B. R. T.Biomass Burning—A Review of Organic Tracers for Smoke from Incomplete CombustionApplied Geochemistry200217129–162
- Stone E. A.Snyder D. C.Sheesley R. J.Sullivan A. P.Weber R. J.Schauer J. J.Source Apportionment of Fine Organic Aerosol in Mexico City During the MILAGRO Experiment 2006Atmos. Chem. Phys.200881249–1259
- Stone E. A.Zhou J. B.Snyder D. C.Rutter A. P.Mieritz M.Schauer J. J.A Comparison of Summertime Secondary Organic Aerosol Source Contributions at Contrasting Urban LocationsEnviron. Sci. & Technol.2009433448–3454
- Strader R.Lurmann F.Pandis S. N.Evaluation of Secondary Organic Aerosol Formation in WinterAtmos. Environ.1999334849–4863
- Subramanian R.Donahue N. M.Bernardo-Bricker A.Rogge W. F.Robinson A. L.Contribution of Motor Vehicle Emissions to Organic Carbon and Fine Particle Mass in Pittsburgh, Pennsylvania: Effects of Varying Source Profiles and Seasonal Trends in Ambient Marker ConcentrationsAtmos. Environ.2006408002–8019
- Subramanian R.Donahue N. M.Bernardo-Bricker A.Rogge W. F.Robinson A. L.Insights into the Primary-Secondary and Regional-Local Contributions to Organic Aerosol and PM2.5 Mass in Pittsburgh, PennsylvaniaAtmos. Environ.2007417414–7433
- Szidat S.Jenk T. M.Gaggeler H. W.Synal H. A.Fisseha R.Baltensperger U.Kalberer M.Samburova V.Wacker L.Saurer M.Schwikowski M.Hajdas I.Source Apportionment of Aerosols by C-14 Measurements in Different Carbonaceous Particle FractionsUniv. Arizona Dept. Geosciences2004475–484
- Turpin B. J.Huntzicker J. J.Secondary Formation of Organic Aerosol in the Los-Angeles Basin—A Descriptive Analysis of Organic and Elemental Carbon ConcentrationsAtmos. Environ. Part a—General Topics199125207–215
- Turpin B. J.Huntzicker J. J.Identification of Secondary Organic Aerosol Episodes and Quantitation of Primary and Secondary Organic Aerosol Concentrations during SCAQSAtmos. Environ.1995293527–3544
- Turpin B. J.Huntzicker J. J.Larson S. M.Cass G. R.Los Angeles Summer Midday Particulate Carbon—Primary and Secondary AerosolEnviron. Sci. & Technol.1991251788–1793
- Wangersky P. J.Dissolved Organic-Carbon Methods—A Critical-ReviewMarine Chemistry19934161–74
- Watson J. G.Cooper J. A.Huntzicker J. J.The Effective Variance Weighting for Least–Squares Calculations Applied to the Mass Balance Receptor ModelAtmos. Environ.1984181347–1355
- Weber R. J.Sullivan A. P.Peltier R. E.Russell A.Yan B.Zheng M.de Gouw J.Warneke C.Brock C.Holloway J. S.Atlas E. L.Edgerton E.A Study of Secondary Organic Aerosol Formation in the Anthropogenic–Influenced Southeastern United StatesJ. Geophys. Res.—Atmospheres2007112
- Yu S. C.Dennis R. L.Bhave P. V.Eder B. K.Primary and Secondary Organic Aerosols Over the United States: Estimates on the Basis of Observed Organic Carbon (OC) and Elemental Carbon (EC), and Air Quality Modeled Primary OC/EC RatiosAtmos. Environ.2004385257–5268
- Zheng M.Cass G. R.Schauer J. J.Edgerton E. S.Source Apportionment of PM2.5 in the Southeastern United States Using Solvent-Extractable Organic Compounds as TracersEnviron. Sci. & Technol.2002362361–2371