
EDITOR:
Measuring the composition of atmospheric aerosols is a challenging problem because of the presence of a wide variety of both inorganic and organic compounds as well as varying volatility, phase, and water content. The Aerodyne Aerosol Mass Spectrometer (AMS) has made significant contributions to our knowledge of aerosols. In Murphy (Citation2016), I pointed out that the framework used for understanding the sensitivity of the AMS to various species did not include the effects of molecular weight and thermal decomposition during vaporization. A comment by Jimenez et al. (Citation2016) supports the accuracy of the AMS and questions the relevance of these effects to the sensitivity of the AMS.
Murphy (Citation2016) and Jimenez et al. (Citation2016) agree on some substantial points. We agree that the time spent by various molecules in the ionization region affects the sensitivity to those molecules. We agree on the need for additional calibrations. There is also agreement that the great majority of AMS data for ambient conditions do not need to be revised by factors of 2 or 3. In discussing why there is no need for such a revision, Jimenez et al. seem to have misunderstood Murphy (Citation2016), in which I stated that the data are unlikely to be off by such factors.
It remains important to recognize the importance of molecular weight and thermal decomposition. In free molecular flow, heavier molecules spend more time in the ion source, increasing the chance of ionization and the sensitivity to those molecules. The molecular weight effect is modulated by thermal decomposition because, for many organics, the species that evaporate off of a 600 C vaporizer have smaller molecular weights than the original molecule.
shows the expected range of sensitivity relative to nitrate. Depending on the amount of thermal decomposition, the sensitivity of the AMS to a given molecule can be anywhere in the shaded region. One can see why the standard AMS organic relative sensitivity of 1.4 (horizontal line) works reasonably well: it is an approximation for species that partially thermally decompose on the vaporizer. That many organics partially decompose is known not only from the overall sensitivity but also from vacuum ultraviolet ionization measurements of CO2 and other fragment molecules (Canagaratna et al. Citation2015).
Figure 1. Predicted pattern of relative sensitivity in the AMS. For moderately high molecular weights, thermal decomposition can change the sensitivity by more than a factor of 3 (vertical arrow). The horizontal line is the standard AMS organic relative sensitivity of 1.4.
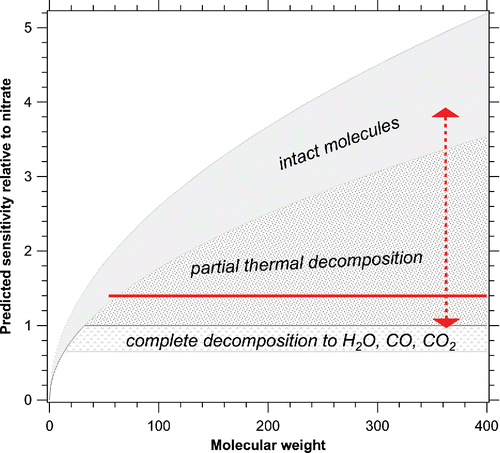
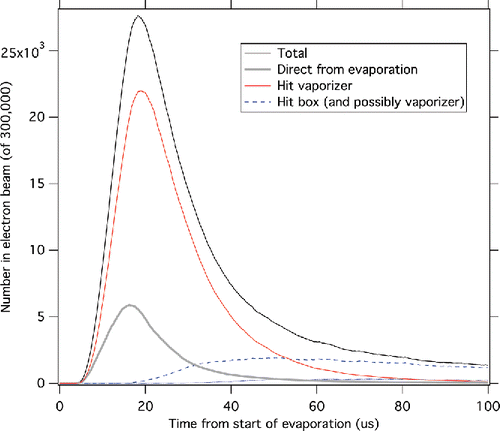
Figure A1. Example event shape for free molecular flow. Conditions are evaporation in a 20-µs-long square pulse, a molecular weight of 80, and temperatures of 300°C for the evaporating particle, 600°C for the vaporizer, and 200°C for the four-sided box around the ion source. Most neutral molecules have at least one wall collision before entering the electron beam.
Besides providing a context for the standard sensitivity, makes two predictions that should be recognized. First, the range of possible sensitivities is quite large and therefore it is possible, even likely, that some classes of organic aerosol, especially in the laboratory or primary emissions, will have significantly different sensitivities than the standard approximation. Second, thermal decomposition has a large influence on sensitivity. This makes it important to systematically calibrate whether or not the thermal decomposition of organics depends on particle properties such as size, oxygen to carbon ratio, what inorganics are present, or other factors.
In their comment, Jimenez et al. (Citation2016) suggest the use of a more empirical approach to AMS calibrations. Indeed, thermal decomposition on the vaporizer is difficult to predict from first principles, making calibrations especially important. Hopefully, the comment by Jimenez et al. will be the start of routine publication of calibrations by many AMS users for the specific types of compounds they are analyzing, rather than the use of a single number for organic sensitivity.
Specific objections by Jimenez et al.
Jimenez et al. (Citation2016) have a number of specific objections to the molecular weight dependence described in Murphy (Citation2016). First, they suggest that the lengths and temperature dependence of pulses of ions from single-particle events are not consistent with Murphy (Citation2016). They do not give an alternative model for the event lengths. There are several difficulties with their arguments: the temperature dependence does not properly represent the model in Murphy (Citation2016), the effect they are looking for may be beyond the resolution of their measurements, and there are multiple temperatures in the system. A full response is quite detailed and is given in Appendix A.
It is important to realize that event lengths are a diagnostic of the physics in the ion source but are not directly related to the AMS sensitivity. The event length is not the same as the time each molecule spends in the ion source. Unless the event length is extremely slow (e.g., minutes), the AMS sensitivity does not depend on the amount of time it takes a particle to evaporate—a positive feature of the AMS. Nor does the event length directly affect the dependence on molecular weight. As long as they are at the same temperature, lighter molecules spend less time in the ion source than heavier molecules for both shorter and longer single-particle events.
Jimenez et al. (Citation2016) are confident that ammonium salts such as ammonium nitrate and ammonium sulfate volatilize to produce ammonia gas and that therefore the sensitivity to ammonium raises questions about the appropriateness of molecular flow. They point out that the AMS has essentially identical sensitivity to the ammonium in ammonium nitrate or ammonium sulfate. The sensitivity in both cases is about a factor of 4 higher than expected from the electron impact cross-section of ammonia gas (Figure 2 of Jimenez et al., Citation2016). Jimenez et al. do not offer a hypothesis for the high sensitivity of the AMS to ammonium.
In Murphy (Citation2016), I offered the hypothesis that if the ammonium compounds evaporate to larger neutral molecules than ammonia gas, then a combination of slower speed and preferential fragmentation of those molecules in the electron beam could produce the observed sensitivity to ammonium. I do not regard the hypothesis of a larger neutral molecule as proven but rather am unable to find any other good explanation for the sensitivity to ammonium, especially any explanation that would not degrade the rest of the mass spectrum. Appendix B discusses detailed reasons and another distant possibility, as well as the sensitivity to water and polycyclic aromatic hydrocarbons (PAHs).
There are some experimental data on the evaporation of ammonium nitrate in vacuum that I was not previously aware of. According to Hildenbrand et al. (Citation2010a,Citationb), “examination of the vapor by effusion-beam mass spectrometry clearly showed the presence of molecular NH4NO3, along with NH3 and HNO3.” So, perhaps, both are present in the AMS. It is not easy to predict the relative amounts in the AMS because, compared to bulk experiments, the AMS has high temperatures that favor decomposition but very fast evaporation that gives little time for that decomposition to occur. Ammonium chloride has been calculated to sublimate as the NH3.HCl complex, then rapidly dissociate (Zhu et al. Citation2007).
The sensitivity to ammonium is measured, so its importance is primarily as a diagnostic of the processes in the AMS ion source. When the sensitivity to ammonium is quantitatively understood, there will be more confidence in the quantitation of other molecules.
Discussion
Jimenez et al. (Citation2016) provide examples with good agreement between organic mass measured by the AMS and other techniques. I agree that enough AMS measurements have been made that, if they consistently had a bias as large as a factor of 2, it would probably have been seen in the many AMS field and laboratory activities. The main issue is having a correct framework so that one can anticipate when the use of a standard sensitivity is inappropriate. The appropriate sensitivity is not easy to determine from instrument comparisons because there is no “gold standard” measurement for aerosol organics and because the combined instrument uncertainties can become large. For example, Figure 3d in Jimenez et al. shows a comparison to another method as the ratio of primary to secondary organics changes. The curve for a factor of 2 in the relative sensitivity of primary and secondary organics is entirely within the combined instrumental uncertainty. If the uncertainty in their figure were centered on the measurements, as is usual, instead of the 1.0 line, then the uncertainty region would include the curve for a factor of 3 in relative sensitivity.
The importance of using a composition-dependent sensitivity depends on the application. Major patterns such as the well-known pie chart map in Zhang et al. (Citation2007) are not going to be significantly affected. A detailed mass balance of aerosol condensation and evaporation as organics oxidize downwind of a source might be more susceptible to changes in sensitivity with organic composition and should be confirmed with other instruments. Laboratory measurements that compare the chemical evolution of specific organic compounds could be even more affected by differing sensitivities for different compounds.
Jimenez et al. (Citation2016) state that the electron impact cross-section arguments in Jimenez et al. (Citation2003) were used to highlight that the ion signals are proportional to mass. A main point of Murphy (Citation2016) is that, when considering specific molecules after evaporation, the ion signals are not proportional to mass but rather mass to the 3/2 power. That the overall organic ion signals are usually approximately proportional to mass is because of thermal decomposition on the vaporizer, a distinction that was not previously recognized. Jimenez et al. (Citation2016) also state that Murphy (Citation2016) was inaccurate about the basis of the AMS sensitivity. In Murphy (Citation2016), I followed the published literature, which includes a theoretical basis for sensitivity (Jimenez et al. Citation2003), but no systematic calibration data. The 2003 paper mentions calibrations as the subject of future work, but they were never published. In this regard, a very positive outcome of the publication of Jimenez et al. (Citation2016) is their Figure 2b, so that now a subset of organic calibrations is published, albeit without a description of the full experiments.
It is also good that the data are shown on a framework to understand them. One can then start to discuss why ammonium appears to be an outlier and whether or not the organic compounds with high sensitivities that were excluded from the overall organic sensitivity (their Figure 2a) are important to other measurements. By personal communications and on the AMS user group presentation web site, there are unpublished calibrations of a number of other organic compounds. Some imply relative sensitivities close to 1.4 and others are much higher (>4). Both my model framework and the unpublished work underscore the importance using a sensitivity appropriate to each type of organic aerosol in the AMS rather than a single number.
Related Research Data
References
- Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildenbrandt, L. Ruiz, Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R. (2015). Elemental Ratio Measurements of Organic Compounds Using Aerosol Mass Spectrometry: Characterization, Improved Calibration, and Implications. Atmos. Chem. Phys., 15:253–272.
- Chien, W.-M., Chandra, D., Lau, K. H., Hildenbrand, D. L., and Helmy, A. M. (2010). The Vaporization of NH4NO3. J. Chem. Thermodynamics, 42:846–851.
- Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R. (2005). A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS)—Instrument Description and First Field Deployment. Aerosol Sci. Technol., 39:637–658.
- Drewnick, F., Diesch, J.-M., Faber, P., and Borrmann, S. (2015). Aerosol Mass Spectrometry: Particle–Vaporizer Interactions and Their Consequences for the Measurements. Atmos. Meas. Tech., 8:3811–3830.
- Dzepina, K., Arey, J., Marr, L. C., Worsnop, D. R., Salcedo, D., Zhang, Q., Onasch, T. B., Molina, L. T., Molina, M. J., and Jimenez, J. L. (2007). Detection of Particle-Phase Polycyclic Aromatic Hydrocarbons in Mexico City Using an Aerosol Mass Spectrometer. Int. J. Mass Spectrom., 263:152–170, and supplemental material.
- Feil, S., Bacher, A., Zangerl, M., Schustereder, W., Gluch, K., and Scheier, P. (2004). Discrimination Effects for Ions with Initial Kinetic Energy Produced by Electron Ionization of C2H2 in a Nier-Type Ion Source. Int. J. Mass Spectrom., 233:325–333.
- Gluch, K., Scheier, P., Schustereder, W., Tepnual, T., Feketeova, L., Mair, C., Matt-Leubner, S., Stamatovic, A., and Märk, T. D. (2003). Cross Sections and Ion Kinetic Energies for Electron Impact Ionization of CH4. Int. J. Mass Spectrom., 228:307–320.
- Hildenbrand, D. L., Lau, K. H., and Chandra, D. (2010a). Thermochemistry of Gaseous Ammonium Nitrate, NH4NO3(g). J. Phys. Chem. B, 114:330–332.
- Hildenbrand, D. L., Lau, K. H., and Chandra, D. (2010b). Revised Thermochemistry of Gaseous Ammonium Nitrate, NH4NO3(g). J. Phys. Chem. A, 114:11654–11655.
- Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X., Smth, K. A., Morris, J. W., and Davidovits, P. (2003). Ambient Aerosol Sampling Using the Aerodyne Aerosol Mass Spectrometer. J. Geophys. Res., 108(D7):8425. doi:10.1029/2001JD001213.
- Jimenez, J. L., Canagaratna, M. R., Drewnick, F., Allan, J. D., Alfarra, M. R., Middlebrook, A. M., Slowik, J. G., Zhang, Q., Coe, H., Jayne, J. T., and Worsnop, D. R. (2016). Comment on “The Effects of Molecular Weight and Thermal Decomposition on the Sensitivity of a Thermal Desorption Aerosol Mass Spectrometer”. Aerosol Sci. Technol., 50(9):i–xv.
- Lampe, F. W., Franklin, J. L., and Field, F. H. (1957). Cross Sections for Ionization by Electrons. J. Am. Chem. Soc., 79:6129–6132.
- Murphy, D. M. (2016). The Effects of Molecular Weight and Thermal Decomposition on the Sensitivity of a Thermal Desorption Aerosol Mass Spectrometer. Aerosol Sci. Technol., 50:118–125.
- Oberheide, J., Wilhelms, P., and Zimmer, M. (1997). New Results on the Absolute Ion Detection Efficiencies of a Microchannel Plate. Meas. Sci. Technol., 8:351–354.
- Poll, H. U., Winkler, C., Margreiter, D., Grill, V., and Märk, T. D. (1992). Discrimination Effects for Ions with High Initial Kinetic Energy in a Nier-Type Ion Source and Partial and Total Electron Ionization Cross-Sections of CF4. Int. J. Mass Spectrom. Ion Proc., 112:1–17.
- Rader, D. J., Trott, W. M., Torczynski, J. R., Castañeda, J. N., and Grasser, T. W. (2005). Measurements of Thermal Accomodation Coefficients. Sandia Report SAND2005-6084.
- Vacher, J. R., Jorand, F., Blin-Simiand, N., and Pasquiers, S. (2007). Electron Impact Ionization Cross-Sections of Toluene. Chem. Phys. Lett., 434:188–193.
- Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., De-Carlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R. (2007). Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes. Geophys. Res. Lett., 34:L13801, doi:10.11029/12007GL029979.
- Zhu, R. S., Wang, J. H., and Lin, M. C. (2007). Sublimation of Ammonium Salts: A Mechanism Revealed by a First-Principles Study of the NH4Cl System. J. Phys. Chem. C., 111:13831–13838.
Appendix
Measured and modeled event lengths
The curves in Figures 1a–c in Jimenez et al. (Citation2016) should be labeled “square root of temperature” rather than “Murphy Citation2016,” since according to Murphy (Citation2016) the square root of temperature dependence is less important than possible changes in molecular weight due to changing thermal decomposition with temperature. Because of the competition between evaporation and decomposition, the net effect of increasing the vaporizer temperature on decomposition is not necessarily a steady increase of decomposition with temperature.
The remaining panels of Figure 1 in Jimenez et al. (Citation2016) compare measured event lengths to a model intended to show the dependence on the square root of mass and the general pulse shape, not the exact event length. Murphy (Citation2016) used a nominal evaporation time of 4 µs but noted that for ammonium nitrate it was somewhere between about 0.5 and 25 µs. With a 15–20 µs evaporation time for ammonium nitrate and a more realistic geometry, a free molecular flow model captures most but not all of the variation in event length.
Both the measured and modeled event lengths are quite complicated. The Drewnick et al. (Citation2015) measurements of event lengths were made with ions pulsed into the time-of-flight (TOF) drift region about every 12 µs. Most of their event lengths are about 30 µs (full width half maximum, FWHM). A 20–30% change is therefore a change of about 6–9 µs, which is difficult to measure with a 12-µs sampling period.
When sampled every 12 µs, there are only two or three high points in a 20–40 µs pulse. Particles arrive at the vaporizer at random times so that the measured points are not synchronized with the arrival time. Those two or three points might hit or miss the peak of the pulse. Furthermore, the data from any single particle are noisy, so many particles must be averaged after aligning the pulses in time.
With averaging it is possible to measure an event length FWHM to somewhat better than the 12-µs interval but, not surprisingly, the results are quite sensitive to the details of the pulse shape, the noise, and the algorithm when the desired accuracy is better than the sample interval. Drewnick et al. aligned the events using the first point above half maximum (personal communication, 2016). This works better than aligning the maxima, which tends to artificially narrow noisy peaks by aligning the noise. Using the rising edge, the derived FWHM are biased high by an amount that depends on the noise and the shape of the pulse. The reason is that the time alignment is not perfect and averaging over many particles smears the pulses by a fraction of the sample interval. An estimate based on some model pulses is that the derived FWHM in Jimenez et al. (Citation2016) are possibly biased high by about 4 µs with an uncertainty of perhaps ±3 to ±6 µs. The accuracy of the derived FWHM gets rapidly worse if there are fewer than roughly 100 ions in each event.
The derived rise times are less accurate than the derived FWHMs. Naturally, one cannot measure the rise time to less than one sample interval. What is not so obvious is that asynchronous sampling often picks up one point before the sampled maximum (which may not be the actual maximum). The sampled rise time is then two sample intervals, even when the actual rise time is much shorter. The rise time measurement of ∼24 µs on the left side of Figure 1d in Jimenez et al. (Citation2016) is almost certainly an artifact of their sampling and averaging procedure.
The modeled event pulses are also complicated. Since Murphy (Citation2016), I have updated the model of free molecular flow through the ion source to make it more accurate, although I want to emphasize that it is still qualitative in some important respects. The most important update is the addition of the four-sided box around the electron beam. Other changes are slight changes in vaporizer dimensions, an improved representation of a cosine distribution of directions from the sloped surface of the vaporizer, an increased vaporizer temperature, and the option of making the temperature of the initial evaporation lower than the temperature of the vaporizer. Some of these changes reduce the FWHM and some increase it, with an overall increase. The ion source box adds considerably to the tail of the pulse profile. Some important aspects that are still uncertain are: possible adsorption of molecules on vaporizer or ion source surfaces, the size and position of the zone defined by the electron beam along with the requirement that any ions formed can reach the detector, the temperature of the initial evaporation, the temperature of the ion source box walls, a small contribution from the time it takes ions to be extracted from the ion source, and the time spent in pores of the vaporizer. In addition to the uncertainties in the physical model, the time it takes a particle to evaporate is poorly constrained. There may be more than one type of nitrate-containing molecule evaporating (Chien et al. Citation2010).
The model shows that there is no single process responsible for the measured event lengths. For example, duplicating the widths given in Jimenez et al. (Citation2016) for ammonium nitrate in the current model suggests contributions of about 15–20 µs each to the FWHM from evaporation and molecular transit time. Surface reactions along with adsorption are one plausible explanation for why ions with mass 30 have a longer event length than other ions from ammonium nitrate (Drewnick et al. Citation2015). Another plausible explanation is differential evaporation and decomposition whereby the last bit of a particle to evaporate could have a different composition and/or has undergone more decomposition than what evaporates initially. Either adsorption or differential evaporation could increase the FWHM but not change the basic molecular weight dependence.
Figure A1 shows a sample evaporation event profile. Most molecules hit a surface before reaching the electron beam. One feature to note is that the leading edge is influenced by molecules that never hit the vaporizer and the trailing edge is strongly influenced by molecules that have hit the walls of the ion source. This means that the width of the pulse will be influenced by molecules that may not be at the temperature of the vaporizer. If so, a simple square root of temperature relationship will not describe the temperature dependence.
Sensitivity to ammonium, water, and PAHs
In the AMS, ammonium has a sensitivity about a factor of 4 larger than expected if it evaporates as ammonia gas (Murphy Citation2016; Jimenez et al. Citation2016). This sensitivity brings up a number of issues about what determines mass spectrometer sensitivity. Electron impact cross-sections are unlikely to explain the sensitivity. The electron impact ionization cross-section of ammonia has been measured (Lampe et al. Citation1957). As far as I can tell, the cross-section of nitric acid has not been measured but the estimated cross-section is unlikely to be off by such a large factor. Furthermore, if it were off, it would affect not just ammonium but all AMS calibrations because the sensitivities for sulfate, organics, and other species are referenced to the NO+ and NO2+ ions from nitrate.
Temperature variations are also unlikely to explain the observed sensitivity to ammonium. Even if different molecules have different translational temperatures, those temperatures would have to be more than 300 K apart to produce even a 20% change in sensitivity. Jimenez et al. (Citation2016) mention possible low thermal accommodation coefficients. Although thermal accommodation coefficients <0.1 can occur in situations such as helium atoms hitting an atomically smooth crystal face, the coefficients for more common situations are larger, such as ∼0.8 for argon or nitrogen on polished stainless steel (Rader et al. Citation2005). In any case, the effect of thermal accommodation coefficients is limited to ∼20% by the temperature span in the ion source.
I cannot see how any sort of bulk flow behavior could explain the sensitivity to ammonium. For reasonable evaporation rates, the evaporating molecules are immediately in free molecular flow. Even for a relatively short 5-µs evaporation time, the first molecules to evaporate have moved over a millimeter by the time the last molecules evaporate. The density resulting from evaporating a single submicron particle into a ∼1 mm3 volume is in the free molecular flow range by orders of magnitude. Particles would have to evaporate in ≪1 µs for significant flow interactions. But even if there were bulk flow with nitric acid and ammonia molecules traveling at the same speed, that still leaves about a factor of 2 in sensitivity to explain. There would have to be something like a plume of ammonia molecules going through the electron beam while many of the nitric acid molecules miss it. Finally, to explain equal sensitivity, any bulk flow or plume would have to form in the same way from both ammonium nitrate and sulfate and for particles of different sizes.
Some AMS users have mentioned to me the possibility of mass discrimination as a possible cause of the high sensitivity to ammonium. One can distinguish distinct types of mass discrimination: ion trajectories that hit or miss the detector, extraction of ions from the source, and variations in detector gain. I will consider these in turn.
It is important to realize that ion trajectories that differ by mass can only occur in certain parts of a mass spectrometer. For constant electric fields, the equations of motion for the trajectories of ions depend only on energy, not mass. If an ion of mass 15 reaches the detector, then an ion with mass 150 starting at the same potential and position will also reach the detector. They will take different amounts of time but always the same trajectories. Having mass discrimination in trajectories requires time-dependent electric fields. (A well-known example is that a quadrupole mass spectrometer uses radio frequency fields to allow only certain masses to reach the detector.) The requirement for time-dependent fields means that the only place there can be trajectory mass discrimination in a simple (that is, no RF lenses or collision cells) TOF spectrometer is in the pulsed extraction region. So for mass discrimination in the spectrometer to explain the sensitivity to ammonia, the pulsed extraction region in the TOF would coincidentally have to have similar mass discrimination to the quadrupole. This would be in addition to a dependence of TOF duty cycle on mass that is accounted for in software (Drewnick et al. Citation2005). Furthermore, a factor of about 4 in mass discrimination between ammonia ions and NO2+ would almost inevitably mean large mass discrimination at higher masses as well and the sensitivity to sulfate and organics would be affected.
The ion source is common to both the quadrupole and TOF versions of the AMS so it is a place to look for a common reason for the sensitivity to ammonium. Simple mass discrimination cannot occur in the ion source for the reason just mentioned: the ion source has constant electric fields and so ions of different masses are extracted with the same efficiency as long as they have the same kinetic energy. The kinetic energy requirement opens a window for discrimination between different fragment ions. The AMS has weak fields to draw the ions out of the source and its high electron beam current implies a potential well for positive ions. In this situation the extraction efficiencies for various ions can be very sensitive to their distributions of initial kinetic energies (Poll et al. Citation1992; Gluch et al. Citation2003; Feil et al. Citation2004). Fragment ions can have a larger range of initial kinetic energies than parent ions because of a requirement for momentum conservation. A quantitative description of extraction of ions from the AMS source is far beyond the scope of this reply. But it is difficult to see how this mechanism could enhance the sensitivity to ammonia and still be consistent with other species and fragmentation patterns.
Microchannel plate (MCP) detectors exhibit mass discrimination, but that is unlikely to explain all of the sensitivity to ammonium. Rather than ion counting, the AMS uses a mostly analog ion measurement that is proportional to the gain of the MCP. Oberheide et al. (Citation1997) reported that a chevron MCP at 850 V per stage had about 15% higher gain at mass 20 than mass 40 for ions with a few kiloelectronvolts of energy. Operation at a higher MCP voltage would reduce this difference because the second MCP stage would be driven further into saturation. If there were a factor of 4 in MCP gain between masses near 17 and 46 it would imply large biases at higher masses as well. A modest reduction in MCP gain at high ion masses could reduce the sensitivity to PAHs and other molecules that produce high mass ions.
At this point, I do not see any mechanism for the high and nearly equal sensitivity to ammonium in ammonium nitrate and sulfate that does not require at least one coincidence to explain this equality. If the larger neutral molecule hypothesis is correct, then those molecules would have to fragment during ionization in just the right way to give the same sensitivity to the ammonium in both ammonium nitrate and sulfate. If the ammonium peaks come from ammonia gas, then whatever mechanism enhances the sensitivity to ammonia gas would coincidentally have to operate equally for ammonium nitrate and sulfate and in the quadrupole and TOF versions of the AMS.
In summary, there are a number of mechanisms that could introduce a mass-dependent sensitivity by amounts of up to 10 or 20%. Examples are a change in the gain of the MCP with ion mass or perhaps different translational temperatures for different neutral molecules. The kinetic energy of the initial ions could conceivably be a larger effect but there is little evidence for it. It is very difficult to find any mechanism that would enhance the sensitivity to ammonia gas by a large amount without also degrading the information in the rest of the mass spectrum. Of the mechanisms I can think of, only the larger ammonium-containing molecule hypothesis does not affect the rest of the spectrum.
Jimenez et al. (Citation2016) suggest that the AMS sensitivity to water also raises questions about the arguments in Murphy (Citation2016). Water is very difficult to calibrate and interpret because its rapid evaporation means that the amount of water in a particle after passage through the inlet and vacuum is extremely uncertain. Jimenez et al. (Citation2016) obtain a high sensitivity to water after making a factor-of-5 correction for evaporation. Given the uncertainties, I do not believe that the sensitivity to water is an argument against free molecular flow.
Jimenez et al. (Citation2016) also argue that the relative sensitivity to certain polycyclic aromatic hydrocarbons (PAHs) is lower than expected from Murphy (Citation2016). They quote a value of 3.2 from Figure 3 in Murphy (Citation2016), which is a misreading of the figure. PAHs have a very low fraction of hydrogen and therefore a low number of electrons per unit mass. Their electron impact cross-sections are therefore expected to be smaller than other organic molecules of similar mass (Jimenez et al. Citation2003). I am not aware of measured electron impact cross-sections for PAHs but toluene (Vacher et al. Citation2007) follows the trend for its hydrogen content. The electron impact cross-sections for PAHs would imply a sensitivity below the pattern of intact molecules with low hydrogen content in my figure, or less than about 2.6 for the molecular weight of the PAHs used by Dzepina et al. (Citation2007). Any thermal decomposition would reduce the sensitivity. PAH sensitivity may also depend on detector performance. It is relevant that PAHs have a sensitivity in the AMS that is higher than many other organics. Because of the small PAH cross-sections, a theory that did not consider molecular weight would predict a lower sensitivity, not higher. That the relative sensitivity to PAHs is higher than many other organics supports the importance of molecular weight.