
ABSTRACT
In this study, we developed an optical monitor to measure light absorption from particulate matter (PM) at 532 nm using a differential photoacoustic absorption spectroscopic (DPAS) technique. The dual-cell system is capable of measuring the photoacoustic signals due to light absorption of total PM and gaseous samples and that of gaseous samples, separately. The resulting differential photoacoustic signal can be used to determine the light absorption purely from the PM species. This measurement method eliminates the interferences from the light-absorbing gaseous species as well as the surrounding low-frequency background acoustic noises. Photoacoustic signals of the DPAS monitor were calibrated with the NO2 gas standards, varying from 100 to 250 parts per billion (ppb). Based on an Allan analysis, a detection sensitivity (2σ) of 0.68 Mm−1 can be achieved in 100 s data acquisition. Using the Jet Burner Test Stand (JBTS) facility at the United Technologies Research Center (UTRC), we measured light absorption by the soot emissions from a representative high-temperature and high-pressure test combustor for aircraft auxiliary power units (APU). The DPAS measurement results at 532 nm, under the high gaseous NO2 conditions, were then compared to the determination of soot mass concentrations from a commercial AVL Micro Soot Sensor (MSS). An excellent linear correlation between the measurements from two instruments was observed. The mass absorption coefficient (MAC) of the soot using the two data sets was 7.4 ± 1.3 m2g−1, in good agreement with the previously reported 8.1 ± 1.7 m2g−1 and the expected value of 7.6 ± 0.6 m2g−1.
Copyright © 2017 American Association for Aerosol Research
EDITOR:
Introduction
It has been well known that atmospheric particulate matters (PM) have significant contribution to the global radiation balance (Stier et al. Citation2007; Carslow et al. Citation2010). Precise measurements of the optical properties of atmospheric aerosol particles are imperative to our understanding of their contributions to global climate change. Light extinction (absorption + scattering) measurements of atmospheric aerosols and particles in the spectral range from infrared (IR) to ultraviolet (UV) have commonly been employed to further our understanding. These properties have been measured in real-time using a number of advanced instruments (Heintzenberg and Charlson Citation1996; Redmond et al. Citation2010), but there are several limitations on the real-time light absorption measurement of PM.
The photoacoustic effect is the physical process of acoustic signal generation from light-absorbing samples (Bell Citation1881). The fundamental theory behind photoacoustic spectroscopy (PAS) is that radiation energy absorbed by the air-borne samples can be released immediately via a non-radiative relaxation process. During the relaxation, heat is generated in a localized region and it creates an acoustic wave that propagates away from the source. The unique characteristics of the PAS technique include high sensitivity of detection, small sample volume, and simplicity of the device. These advantages can only be exploited when the technique is combined with a suitable laser source, however (Rosengren Citation1975). This method has been widely applied in studying a large variety of chemical and physical phenomena such as forbidden electronic transitions and trace gas detection (West et al. Citation1983; Miklos et al. Citation2001).
Optical properties of atmospheric aerosols including both absorption and scattering have a great deal of variability dependent upon weather, location, and aerosol composition (Bergstrom et al. Citation2007; Li et al. Citation2007; Montilla et al. Citation2011). One of the challenges in atmospheric PM optical absorption measurements comes from the significant interferences in the light absorption due to atmospheric gas species such as nitrogen dioxide (NO2), ozone (O3) and water. The NO2 molecule has a pronounced and broad electronic absorption feature covering the UV-visible spectral region. Its absorption cross sections from UV to near-IR at different temperatures and pressures have been extensively investigated by various spectroscopic techniques at different spectral resolutions (Burrows et al. Citation1998; Voigt et al. Citation2002; Vandaele et al. 2002; Osthoff et al. Citation2006). At standard temperature and pressure conditions (T = 298 K and P = 100 kPa), 10 ppb NO2 in ambient air would lead to an absorption coefficient of 3.6 Mm−1 at 532 nm, calculated from the published absorption cross section of NO2 from the cavity ring-down (CRDS) spectroscopic investigation by Osthoff et al. (Citation2006). O3 shows multiple absorption bands in the entire UV-visible region (Hearn Citation1961; Molina and Molina Citation1986; Brion et al. Citation1998; Bacis et al. Citation1998; Burrows et al. Citation1999). However, transition intensity of the O3 Chappuis band between 375 nm and 650 nm is normally weak (Brion et al. Citation1998), compared to that of NO2. In the UV, or red and near-IR spectral regions, light absorption by atmospheric O3 becomes more important (Burkholder and Talukdar Citation1994; Rothman et al. Citation2005). Thus the commercial soot monitors using photoacoustic techniques such as the AVL Micro Soot Sensor (MSS) are normally operated at the near-infrared spectral region (λ = 880±5 nm) to minimize the impact of atmospheric NO2, O3, and water on optical absorption (Lobo et al. Citation2015). To investigate PM absorption in the spectral range from ultraviolet (UV) to infrared (IR), it is necessary to eliminate these interferences from the gas phase absorption.
In this study, we demonstrated a differential photoacoustic absorption spectrometry (DPAS) technique for the measurement of PM absorption. The constructed system contains a dual-photoacoustic-cell system. Total light absorption from both PM and gaseous species can be measured within the sample cell, while the absorption from the gaseous species alone can be measured within the reference cell. Therefore, the instrument is capable of measuring PM optical absorption in the presence of light absorbing gaseous species, such as NO2, ozone, and water, while still maintaining the fast time response and high sensitivity of the photoacoustic technique.
Experimental
Design of the DPAS instrument
Similar to most PAS PM absorption measurement systems (Arnott et al. Citation2005b; Lack et al. Citation2006; Tian et al. Citation2009; Lack et al. Citation2012; Sharma et al. Citation2013), our DPAS PM absorption monitor consists of a light source, a dual-cell photoacoustic apparatus, a phase sensitive detection system, and data acquisition electronics. A schematic plot of the DPAS instrument is shown in .
Figure 1. Schematic plot of the DPAS PM light absorption measurement technique.
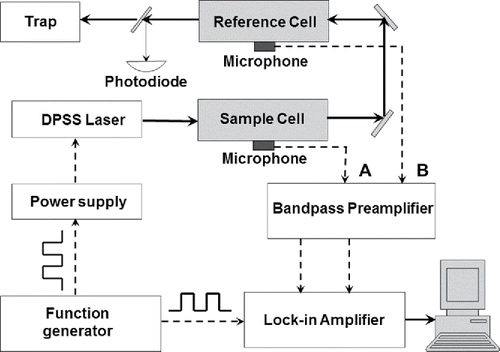
A high-power continuous-wave (cw) diode-pumped solid state (DPSS) laser (MGL-III-532, Opto Engine, LLC.) was used as the light source to provide the radiation power up to 200 mW around 532 nm. Using a high-resolution visible spectrometer, we found the peak frequency of the DPSS laser is 532.24 nm with a full-width-at-half-maximum (FWHM) of about 0.04 nm. The laser output power was modulated at the resonance frequency of the photoacoustic cells via a waveform control on its electrical DC power supply. The control voltage waveform was generated by a custom-made miniature function generation.
The dual-cell photoacoustic apparatus contains two identical cylindrical acoustic resonators: one is used as the sample photoacoustic cell, and the other as the reference photoacoustic cell. A stainless steel Y-splitter divides the primary sample inlet of the system into the two cells. A high efficiency particulate arresting (HEPA) filter was applied at the sample inflow to the reference cell to filter out the PM species. An identical HEPA filter was applied at the sample outflow of the sample cell to balance the small pressure drop between the two flow paths. The HEPA filter manufacturer claims that it comprise fibers that are specific for particulate removal where corrosive gases like NO2 and O3 are to be filtered efficiently as they have excellent chemical resistance. The precise dimension and single-pass configuration of the photoacoustic cells guaranteed that optical path length, resonance frequency, and cavity quality of the two cells are nearly identical. Thus, the microphone signal from the sample photoacoustic cell comes from the total light absorption by both PM and gaseous species in the sample, while the signal from the reference cell is due to the light absorption exclusively by the gaseous species in the sample. The advantage of this design over the conventional single-cell photoacoustic approaches is that the light absorption interferences from the gaseous species such as NO2 and ozone, along with the wall effect from the laser radiation, can be eliminated after the differential data processing. Since PM species like black carbon soot particles can be completely filtered out in the reference cell, the difference between the sample and reference photoacoustic signals is purely due to the PM light absorption.
The microphone detector used for the DPAS device was the SiSonic silicon-based microelectromechanical system (MEMS) microphone (Model# SPU0409HD5H from Knowles). Sensitivity of this MEMS microphone is -42dB at acoustic frequency of 1 kHz and its signal-to-noise ratio (S/N) is 59dB. Given the small dimension of the Knowles MEMS microphone, 2.7 mm × 3.8 mm, we found that a linear-array detector with multiple microphones can be applied as the photoacoustic detector, which produces a two times higher S/N than a single microphone detector. An integrated printed circuit board (PCB) containing an array detector with 4 microphones was designed and constructed for this application. The custom-made PCB is only 5.0 cm × 1.5 cm in dimension. Several band-pass filters and low-noise voltage preamplifiers were also integrated onto the PCB to reduce electronic noise prior to signal processing.
In addition to the microphone array detector, the phase sensitive detection system also includes a custom-made miniature function generator and a commercial analog lock-in amplifier (Model LIA-MV-150 from Femto). Square-wave output from the function generator that matches the resonance frequency of the photoacoustic cells was used to control the electrical DC power supply of the DPSS laser. In the meantime, the TTL reference signal from the same function generator was sent to the lock-in amplifier as the reference input. The AC voltage from the chain of amplifiers was demodulated via the lock-in amplifier. Data acquisition procedure of the DPAS prototype was carried out via a 24-bit resolution DAQ device (Model USB-2404-10 from Measurement Computing), in which an analog-to-digital converter (ADC) was used to convert the analog voltage outputs from the lock-in amplifier into the 24-bit-resolution digital outputs for the computer to record.
The samples including both PM and gaseous species were drawn into the DPAS instrument via a compact diaphragm pump. A stainless steel critical orifice was placed before the pump to control the total sample flow rate. The instrument normally operates at a sample flow rate of 1 liter per minute (LPM). However even with the flow rate increasing to 2 LPM, we did not observe any variation of background noise level. Given the total sample volume of 100 cubic centimeters inside the DPAS instrument, the sample residence time is 6 s at a flow rate of 1 LPM.
DPAS signal calibration and sensitivity analysis
In this study, ppb levels of NO2 were used as the calibration standards and their absorption coefficient at 532 nm was calculated from the published spectroscopic results of NO2 (Osthoff et al. Citation2006). The NO2 gas standards were generated by mixing two flow streams: a dry particle-free nitrogen flow and a flow of 10.0 ± 0.2 parts per million (ppm) NO2 in dry air. Both flows were controlled via the MKS mass flow controllers. The reason dry nitrogen was used in this calibration process is because our main application of the DPAS in this study is to measure aviation soot, which is present at a nearly oxygen-free environment due to the combustion process and high-level of dilution with dry nitrogen, as we discussed in the next section.
After the signal response of the DPAS device was calibrated with the NO2 standards, Allan analysis was performed to determine detection limit and signal stability of the instrument. In this approach, the NO2 flow was switched off. Thus only particle-free dry nitrogen air flowed through the instrument (both the sample and reference cells) at the flowrate of 1.0 LPM. Signal measurements using this scheme spanned a continuous run of many hours. The Allan standard deviation (σ) was then calculated from the obtained time series of measurement signals as a function of integration time.
Instrument evaluation and testing
Instrument evaluation and testing of combustion soot measurement was performed at Jet Burner Test Stand (JBTS) of the United Technologies Research Center (UTRC). The soot source at the JBTS used in this study was from a representative high-temperature and high-pressure test combustor for aircraft APU. It was placed in a vessel that allows inlet air pressures up to 2000 kPa and temperatures up to 500°C. The exhaust samples from the test combustor were extracted at the exit plane (1 m downstream from the combustor) by a 5-nozzle sampling probe. The probe was connected to a three-way stainless steel splitter to enable simultaneous gas sampling, PM sampling, and pressure control, as shown in . The gas sample line (ANNEX 16) was compliant with the International Civil Aviation Organization (ICAO) ANNEX 16 requirements (ICAO Citation2008). The gas sample was a particle-free, undiluted, and heated line that fed gaseous species measurement instruments. The gaseous species measurement included CO2, CO, O2, NOx, and unburned total hydrocarbons. Due to the high-temperature and high-pressure combustion, this type of combustor normally generates a relatively high concentration of NO2 with appreciable soot emissions compared to other internal combustion engines. This study demonstrated that the DPAS instrument is capable of measuring PM light absorption in the presence of high background absorption by gaseous species.
Figure 2. The PM sampling and measurement system at the JBTS facility of UTRC.
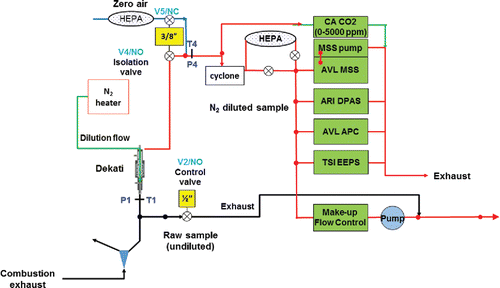
The PM sampling line started with an 8 m long, 8 mm internal diameter (ID) stainless steel tubing whose temperature was maintained at 160°C ± 15°C, in compliance with the AIR6241 specification (AIR6241, Citation2013). The sampling line also had a pressure control valve that regulated the sample flow entering a Dekati diluter. The exhaust sample was diluted with particle-free dry nitrogen in the Dekati diluter by a factor of 10 to suppress new particle formation and condensation of water and other volatile species on the non-volatile soot particles. The sampling line after the Dekati diluter was a 25 m long stainless steel tube (8 mm ID) maintained at 60 ± 15°C. The PM sample flow rate in the PM sampling line was approximately 25 LPM, in agreement with the AIR6421 specification.
The applied PM measurement instrument set included an AVL micro soot sensor (MSS) to measure soot mass concentration, an AVL advanced particle counter (APC) to measure soot number concentration, a TSI engine emission particle sizer (EEPS) to monitor particle size distribution as well as the Aerodyne DPAS monitor to determine soot optical absorption at 532 nm. The EEPS instrument has a time resolution of 1 s, which allows us to distinguish combustion soot and nucleation particles in real time (Johnson et al. Citation2004; Xue et al. Citation2015).
A Y-splitter with a solenoid 3-way valve system was installed on the PM sampling line to deliver the PM sample to the MSS and DPAS for instrument comparison. It created two selections for the PM samples; directly into the MSS and DPAS instruments or passing through a HEPA filter first. Letting the PM sample pass through the HEPA filter (with negligible pressure drop) was considered “zeroing” the instruments.
Results and discussions
Signal calibration with the NO2 standards
The calibration procedure is critical to accuracy of the photoacoustic PM light absorption measurement. Due to lack of a PM calibration standard, PAS calibration are typically performed with light-absorbing gas standards. Arnott et al. (Citation2000) used high-concentration NO2 to calibrate their PAS instrument. Lack et al. (Citation2006) utilized ozone, whose optical absorption levels were measured with the CRDS technique (O'Keefe & Deacon 1988; Scherer et al. Citation1997; Ball and Jones Citation2003). Most recently, Tian et al. (Citation2009) demonstrated a PAS calibration method through simultaneous measurements on aerosol optical absorption and the oxygen A-band absorption at 762 nm. Lewis et al. (Citation2008) and Nakayama et al. (Citation2015) applied the “closure method” to calibrate the photoacoustic signals. This approach simultaneously measures extinction, absorption and scattering from high concentrations of light-absorbing gas or PM standards.
The outputs from the microphone array detectors of the DPAS device were AC signals in voltage, normally in the range of µV to mV. To determine PM light absorption via the DPAS measurements, it is necessary to convert the signal response from voltage into absorption coefficient. A response calibration process is thus required for the DPAS PM light absorption measurement at 532 nm. In this study, we adopted a calibration scheme using the NO2 standards from 100 ppb to 250 ppb in concentration, since the absorption cross section of NO2 at 532 nm has been precisely determined to be (1.45 ± 0.06) × 10−19 cm2 via a CRDS spectroscopic investigation by Osthoff et al. (Citation2006).
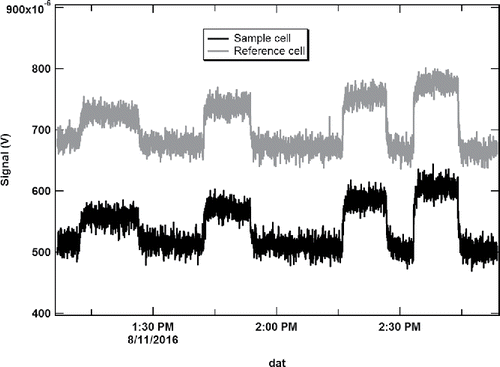
In addition, they concluded that the variation in absorption cross-section for typical sample temperature (15°C to 35°C) and pressure variability (±3 kPa) is negligible. Using this determined absorption cross section value, we were able to convert the voltage outputs from the DPAS instrument at each NO2 concentration into an absorption coefficient at 532 nm in units of Mm−1, to generate the calibration plots for both sample and reference cells. A portion of the measured signal response from both the sample and reference photoacoustic cells, corresponding to the NO2 standards, is shown in . Due to light absorption of the optical windows, which would be at the resonance frequency, there were appreciable background offsets for the sample and reference cells (approximately 500 and 700 μV, respectively). Thus the calibrated signals from the NO2 absorption were determined by subtracting the background offsets from the NO2 measurements.
Figure 3. Signal responses from both sample and reference cells to the NO2 standards.
There are several experimental parameters that influence the accuracy of the applied NO2 calibration method. For instance, variation in resonance frequency could significantly impact the accuracy of signal response for the photoacoustic techniques. Fixed-length cylindrical acoustic resonators were used for this DPAS technique; the acoustic resonance frequency (ω) of the DPAS device can be determined by sound velocity (c) and cell length (l) as the following:(1)
Sound velocity is only dependent on temperature and molar mass of the gas medium, not on pressure. For this reason, the calibration process has to be performed at the same temperature and air composition as the sample measurements. As we will discuss in the section of instrument evaluation and comparison, the combustion soot emissions were highly diluted with dry nitrogen and the sample temperature and pressure was maintained at 20°C and 99.3 kPa. Therefore the signal calibration was carried out in the laboratory at 20°C and 99.3 kPa with dry nitrogen as the carrier gas of the NO2 standards.
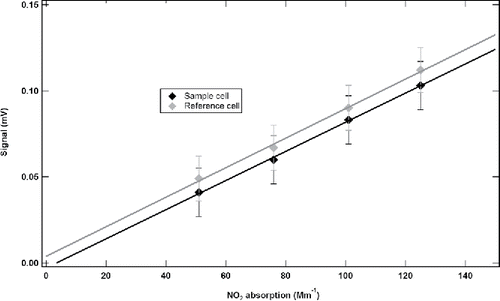
The obtained calibration curves for both sample and reference cells are shown in . The measured DPAS signals were determined by subtracting the background offsets from the measured NO2 signals. The determined uncertainties in y-axis are square root of the sum of the variances from the two measurements, while the uncertainties in x-axis are dependent on the accuracy of the cited NO2 absorption cross section. There was a slight difference in signal offset from the two cells, which may arise from minor variations in the electronic components. The slope of the calibration curves was determined to be 0.00116 ± 0.00003 mV/Mm−1 for the sample cell and 0.00117 ± 0.00005 mV/Mm−1 for the reference cell. They are essentially identical when the experimental uncertainties are taken into account, indicating that the two acoustic cells have the same photoacoustic signal response to the NO2 standards.
Figure 4. Calibration curves of DPAS cells derived from NO2 standards.
Allan analysis and detection limit
Allan analysis is an effective way of analyzing stochastic noises via determination of Allan variance (σ2) over different integration times of measurement signals (Allan Citation1966). Allan analysis has been used as a standard technique to characterize frequency stability and detection sensitivity of many atmospheric measurement instruments (Werle et al. Citation1993; Li et al. Citation2012; Dyroff et al. Citation2014). In such analysis, the calculated Allan variance represents the instrument stability due to random noise rather than systematic errors.
In this study, the DPAS signal measurements for Allan analysis with the phase sensitive detection scheme spanned a continuous run of at least 24 hours after the DPAS measurement became stable. The obtained signals in voltage were converted into absorption coefficient (with the unit of Mm−1) by applying the calibration curves described in the previous section.
We then calculated the Allan variance (σ2) as a function of signal integration time to generate the Allan plot for signal stability analysis. The obtained Allan plot is demonstrated in . The top plot in black is the time series of the raw light absorption measurement from the DPAS instrument with a continuous particle-free nitrogen flow at 1.0 LPM and the bottom plot is the calculated Allan variance as a function of the integration time. The Allan analysis indicates that the maximum stability time of the DPAS instrument is up to 500 seconds.
Figure 5. Allan analysis of the DPAS PM light absorption instrument when measuring pure N2.
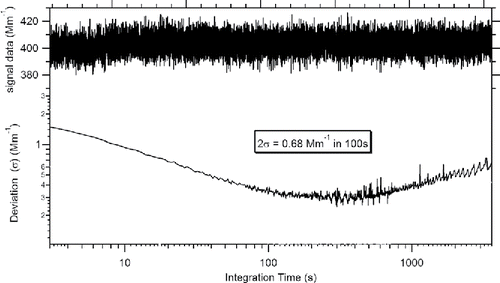
Based on the Allan analysis, a detection sensitivity of 0.68 Mm−1 (2σ) in 100 s can be also achieved for the single acoustic cell measurement. For the differential measurement, the detection limit is 0.95 Mm−1 or less in 100 s data acquisition. In comparison, Sharma et al. (Citation2013) demonstrated a novel multi-wavelength photoacoustic-nephelometer spectrometer that is of a detection limit of 4.4 Mm−1 in 35 s at 542 nm. Nakayama et al. (Citation2015) characterized the three-wavelength photoacoustic soot spectrometer (PASS-3) and reported a detection of 1.9 Mm−1 for 10-minute average at 532 nm. Using a Herriot cell multi-pass configuration, Lack et al. (Citation2006) reported a detection limit of 0.08 Mm−1 in 60 s data acquisition from a single-cell photoacoustic device at 532 nm.
Instrument evaluation and comparison
An important advantage of the DPAS technique over previous PAS PM absorption measurement techniques is the real-time and continuous monitoring of PM absorption even in the presence of strong gas phase absorption. We tested and evaluated this capability by measuring absorption coefficient of soot particles under the appreciable background NO2 levels of the APU test combustor at UTRC. The UTRC JBTS facility provided us four test points in two repeated shifts: high-NO2 and low-soot (#1); low-NO2 and low-soot (#2); low-NO2 and high-soot (#3); high-NO2 and high-soot (#4). In the first shift, the sample probe was broken during the test so we just obtained one test point. The obtained measurement results from the DPAS instrument during the second shift were shown in . The starting time was called by the engine operator when the engine operation and emissions were fully stabilized. The stable time period for each test point was: 0 – 800 for #1, 1000 – 1800 for #2, 2800- 4000 for #3, and 4400–5200 for #4. The rest were transient periods when the engine conditions were being adjusted.
Figure 6. Time series of absorption coefficients from the DPAS measurements when measuring soot + NO2.
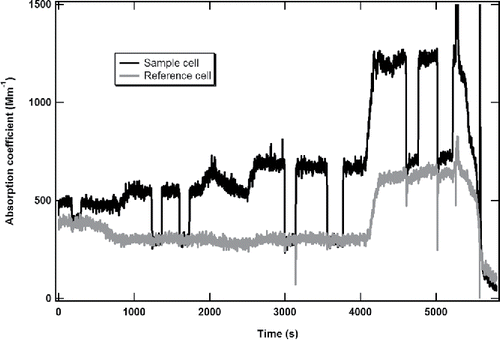
The dark line represents the total absorption signal from the sample cell and the grey line is the gas absorption from the reference cell. As a reference point, the NO2 level at the undiluted Annex 16 line was 7 ppm at 1500 s of the data acquisition time. Given the dilution factor of ten for the AIR 6241 PM sampling line, the NO2 level at the PM line was about 700 ppb, more than 10 times higher than the estimated ground-level NO2 concentrations in North America (Lamsal et al. Citation2008).
Since the background offset of the photoacoustic signal is typically not guaranteed stable for a long period of time, during the measurement, we also performed multiple “signal zeroing” measurements by allowing the PM sample to flow through an additional HEPA filter before entering the DPAS instrument. The zeroing processes were performed for each test point, as shown at the sudden loss of the signals for periods of 2–3 minutes in . The zeros of the instrument from the sample cell are identical to the signal values of the reference cell except with high NO2 concentration, implying there is no absorption due to the PM samples in the reference cell. However for test point #4, we found the zeros from the sample cell were slightly higher than those from the reference cell. As shown in the figure, the soot concentration is also high at that condition. It seems likely that the soot particles accumulated in the HEPA filter are capable of adsorbing the NO2 molecules in the sample flow. This would result in a lower baseline offset in the reference cell than that in the sample cell, as shown in . One potential solution to avoid this problem in the future is to heat the HEPA filter to a higher temperature to reduce NO2 adsorption by the soot particles.
To obtain the optical absorption of the combustor-emitted soot particles from the DPAS measurements, we subtracted the reference cell signal from the sample cell signal to remove the influence of the background NO2. The resulting differential light absorption signal was then compared with the soot concentrations determined by the MSS measurements, as illustrated in . The black line is the time series of the DPAS-measured absorption coefficient of the soot particles at 532 nm and the grey line is the soot mass concentration measured by the MSS at 880±5 nm. The correlation between the two photoacoustic measurements is excellent, indicative of the DPAS capability of measuring PM light absorption in the presence of high concentrations of light-absorbing NO2.
Figure 7. Comparison between soot absorption coefficients from the DPAS measurements and soot mass concentrations from the MSS measurements.
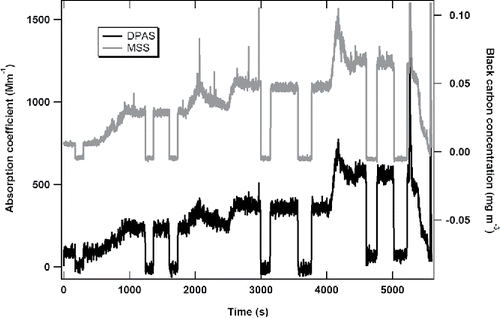
Using the zero measurements, we determined the averaged absorption coefficient and mass concentration of the soot for each test point from the DPAS and MSS measurements. Although the MSS is a single-cell photoacoustic technique, it is calibrated to correlate to elemental carbon (EC) mass concentration via the NIOSH 5040 thermo-optical method (Lobo et al. Citation2015). Thus the MSS measurement reports BC mass concentrations directly, without the determination of PM absorption coefficient and the calculation of mass concentration by applying the mass absorption coefficient (MAC) at 880 nm. Its measurement uncertainty is 17% at the mass concentration of 23 μg/m3.
A linear correlation plot of the averaged results from both the DPAS and MSS measurements is demonstrated in . The obtained correlation coefficient (R2) is 0.999, implying an excellent agreement between the DPAS and MSS instruments. Additionally, this linear plot of soot absorption coefficients from DPAS in inverse megameters (Mm−1) versus the MSS measurement in μg/m3 allows determination of the MAC of 7.4 ± 0.1 m2g−1 at 532 nm for the soot particles emitted from the APU test combustor. The reported uncertainty of the MAC at 532 nm represents only the uncertainty in the linear fit, and not the propagated uncertainty that accounts for the calibration uncertainty. If including the 17% calibration uncertainty from the MSS measurement, the MAC of aviation soot at 532 nm should be 7.4 ± 1.3 m2g−1.
Figure 8. Determination of MAC for the soot particles at 532 nm via the linear correlation between the DPAS and MSS results.
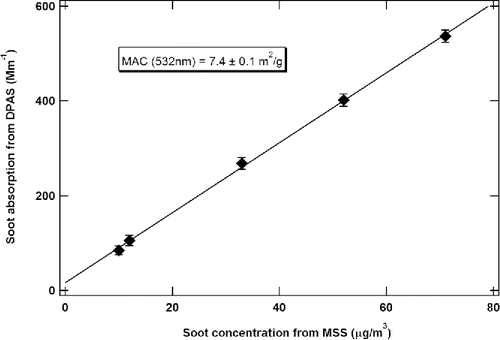
This result is consistent with the previously reported MAC of 8.1 ± 1.7 m2/g by Cross et al. Citation2010. In addition, the generally accepted MAC of aviation soot at 633 nm is 6.4 ± 0.5 m2g−1, determined by Mulholland and Choi (Citation1998). If we assume that the Absorption Angstrom Exponent (AAE) of non-volatile soot particles is 1.0 (Bond and Bergstrom Citation2006; Russell et al. Citation2010), the MAC of aviation soot at 532 nm is expected to be 7.6 ± 0.6 m2g−1, consistent with the determined result in this study.
At present, the atmospheric PM absorption coefficient in the presence of gas phase absorption can be measured by collecting the samples on a filter substrate and measuring light attenuation due to the collected samples. One of the filter-based techniques commonly used is aethalometer (Hansen et al. Citation1982; Weingartner et al. Citation2003; Arnott et al. Citation2005a). The multi-angle absorption photometer (MAAP) technique (Petzold & Schonlinner Citation2004) is another widely used filter-based measurement technique, especially in black carbon emission measurements on internal combustion engines. The filter-based methods usually need various corrections to account for changes in the filter properties due to particle deposit (Cappa et al. Citation2008; Lack et al. Citation2008).
The PAS measurements of PM light absorption are capable of providing real-time data acquisition (Arnott et al. Citation2005b; Lack et al. Citation2012). However, none of these techniques operates with a simultaneous gas phase subtraction to eliminate the interference of light-absorbing gas species in real-time. Filtered-based gas phase corrections are normally applied after a certain period of measurement. During the PAS measurements on urban pollution events, Lack et al. (Citation2012) found that the gas phase absorption at 532 nm contributed on average 10% of total absorption, while at 404 nm it increases to 35%. This observation indicates that the gas phase absorption is appreciable compared to the PM absorption in the green and blue spectral regions. One approach to eliminate the interference from gas absorption is to use a gas scrubber to remove NO2 from the sample (Liu et al. Citation2015). Another method is to measure gas light extinction using cavity ring-down spectrometers (CRDS) and determine light absorption after the theoretical correction of the contribution from Rayleigh scattering (Lack et al. Citation2012). In this study, we demonstrated that the DPAS measurement technique is able to measure PM light absorption at 532 nm in the presence of a significant contribution from the gas phase absorption. Furthermore, this technique can be easily extended into applications at other wavelengths from UV to IR, which can be used to determine the PM absorption due to black carbon and brown carbon, respectively (Moosmüller et al. Citation2009).
Conclusions
We developed a PM light absorption measurement technique based on the differential photoacoustic method. The instrument measures both the light absorption by the whole sample in the sample cell and by the background gas in the reference cell. Absorption coefficient signals of the DPAS device were calibrated with NO2 standards. Using the Jet Burner Test Stand (JBTS) facility at the United Technologies Research Center (UTRC), we demonstrated that this technique could eliminate the interferences from the background gas and low-frequency background acoustic noise. The response of the DPAS device to combustion soot emissions under high NO2 concentrations, was compared to a commercially available micro soot sensor (MSS) and an excellent linear correlation between the two photoacoustic measurement instruments was observed. Based on the Allan variance analysis, a detection sensitivity (2σ) of 0.68 Mm−1 was achieved in 100 s data acquisition.
Acknowledgments
We are grateful for the technical assistance and logistical support offered by the JBTS crew at UTRC during the test combustor study. We also thank Dr. Andy Freedman at Aerodyne Research, Inc., Dr. Andreas Beyersdorf at California State University – San Bernardino, Dr. Richard Moore and Dr. Bruce Anderson at NASA Langley Research Center for helpful discussions.
Funding
The authors acknowledge the financial support of NASA, under Contracts # NNX13CL42P & NNX14CL90C.
References
- Aerospace Information Report (AIR) 6241. (2013). Procedure for the continuous sampling and measurement of non-volatile particle emissions from aircraft turbine engines. SAE International.
- Allan, D.W. (1966). Statistics of Atomic Frequency Standard. Proc. IEEE, 54:221–230.
- Arnott, W. P., Moosmüller, H., and Walker, J. W. (2000). Nitrogen Dioxide and Kerosene-Flame Soot Calibration of Photoacoustic Instruments for Measurement of Light Absorption by Aerosols. Rev. Sci. Instrum., 71:4545–4552.
- Arnott, W. P., Hamasha, K., Moosmüller, H., Sheridan, P. J., and Ogren, J.A. (2005a). Towards Aerosol Light-Absorption Measurements With a 7-Wavelength Aethalometer: Evaluation With a Photoacoutic Instrument and 3-Wavelength Nephelometer. Aerosol Sci. Technol., 39:17–29.
- Arnott, W. P, Zielinska, B., Rogers, C. F., Sagebiel, J., Park, K., Chow, J., Moosmüller, H., and Watson, J. G. (2005b). Evaluation of 1047-nm Photoacoustic Instruments and Photoelectric Aerosol Sensors in Source-Sampling of Black Carbon Aerosol and Particle-Bound PAHs from Gasoline and Diesel Powered Vehicles. Environ. Sci. Technol., 39:5398–5406.
- Bacis, R., Bouvier, A. J., and Flaud, J.-M. (1998). The Ozone Molecule: Electronic Spectroscopy. Spectrochim. Acta. A, 54:17–34.
- Ball, S. M, and Jones, R.L. (2003). Broad-Band Cavity Ring-Down Spectroscopy. Chem. Rev., 103:5239–5262.
- Bell, A. G. (1881). Upon the Production of Sound by Radiant Energy. Philos. Mag., 11:510–528.
- Bergstrom, R. W., Pilewskie, P., Russell, P. B., Redemann, J., Bond, T. C., Quinn, P. K., and Sieran, B. (2007). Spectral Absorption Properties of Atmospheric Aerosols. Atmos. Chem. Phys., 7:5937–5943.
- Bond, T. C., and Bergstrom, R. W. (2006). Light Absorption by Carbonaceous Particles: An Investigative Review. Aerosol Sci. Technol., 40:27–67.
- Brion, J., Chakir, A., Charbonnier, J., Daumont, D., Parisse, C., and Malicet, J. (1998). Absorption Spectra Measurements for the Ozone Molecule in the 350–830 nm Region. J. Atmos. Chem., 30:291–299.
- Burkholder, J. B., and Talukdar, R. K. (1994). Temperature-Dependence of the Ozone Absorption-Spectrum Over the Wavelength Range 410 to 760 nm. Geophys. Res. Lett., 21:581–584.
- Burrows, J. P., Dehn, A., Deters, B., Himmelmann, S., Richter, A., Voigt, S., and Orphal, J. (1998). Atmospheric Remote-Sensing Reference Data from GOME: Part I. Temperature-Dependent Absorption Cross-Sections of NO2 in the 231–794 nm Range. J. Quant. Spectrosc. Radiat. Transfer, 60:1025–1031.
- Burrows, J. P., Richter, A., Dehn, A., Deters, B., Himmelmann, S., Voigt, S., and Orphal, J. (1999). Atmospheric Remote-Sensing Reference Data From GOME: 2. Temperature-Dependent Absorption Cross Sections of O3 in the 231–794 nm Range. J. Quant. Spectrosc. Radiant. Transfer, 61:509–517.
- Cappa, D. C., Lack, D. A., Burkholder, J. B., and Ravishankara, A. R. (2008). Bias in Filter-Based Aerosol Light Absorption Measurements Due to Organic Aerosol Loading: Evidence from Laboratory Measurements. Aerosol Sci. Technol., 42:1022–1032.
- Carslaw, S. K., Boucher, O., Spracklen, V. D., Mann, W. G., Rae, L. J. G., Wordward, S., and Kulmala, M. (2010). A Review of Natural Aerosol Interaction and Feedbacks Within the Earth System. Atmos. Chem. Phys., 10:1701–1737.
- Cross, E. S., Onasch, T. B., Ahern, A., Wrobel, W., Slowik, J. G., Olfert, J., Lack, D. A., Massoli, P., Cappa, C. D., Schwarz, J. P., Spackman, R., Fahey, D. W., Sedlacek, A., Trimborn, A., Jayne, J. T., Freedman, A., Williams, L. R., Ng, N. L., Mazzoleni, C., Dubey, M., Brem, B., Kok, G., Subramanian, R., Freitag, S., Clarke, A., Thornhill, D., Marr, L. C., Kolb, C. E., Worsnop, D. R., and Davidovits, P. (2010). Soot Particle Studies – Instrument Inter-Comparison – Project Overview. Aerosol. Sci. Technol., 44:8, 592–611.
- Dyroff, C., Zahn, A., Sanati, S., Christner, E., Rauthe-Schöch, A., and Schuck, T. J. (2014). Tunable Diode Laser in-situ CH4 Measurements Aboard the CARIBIC Passenger Aircraft: Instrument Performance Assessment. Atmos. Meas. Tech., 7:743–755.
- Hansen, A. D. A., Rosen, H., and Novakov, T. (1982). Real-Time Measurement of the Absorption Coefficient of Aerosol Particles. Appl. Opt., 27:3060–3062.
- Hearn, A. G. (1961). The Absorption of Ozone in the Ultraviolet and Visible Regions of the Spectrum. Proc. Phys. Soc., 78:932–940.
- Heintzenberg, J., and Charlson, J. R. (1996). Design and Applications of the Integrating Nephelometer: A Review. J. Atmos. Ocean Tech., 13:987–1000.
- International Civil Aviation Organization (ICAO). (2008). Environmental Protection: International Standards and Recommended Practices – Annex 16 to the Convention on International Civil Aviation, 3rd ed., Vol.II, Aircraft Engine Emissions, Montreal, QC, Canada.
- Johnson, T., Caldow, R., Pocher, A., Mirme, A., and Kittelson, D. B. (2004). An New Electrical Mobility Particle Sizer Spectrometer for Engine Exhaust Particle Measurements. SAE Technical Paper 2004-01-1341. Society of Automotive Engineers.
- Lack, D. A., Lovejoy, E. R., Baynard, T., Pettersson, A., and Ravishankara, A. R. (2006). Aerosol Absorption Measurement Using Photoacoustic Spectroscopy; Sensitivity, Calibration, and Uncertainty Developments. Aerosol Sci. Technol., 40:697–708.
- Lack, D. A., Cappa, D. C., Covert, S. D., Baynard, T., Massoli, P., Sierau, B., Bates, S. T., Quinn, K. P., Lovejoy, R. E., and Ravishankara, R. A. (2008). Bias in Filter-Based Aerosol Light Absorption Measurements Due to Organic Aerosol Loading: Evidence from Ambient Measurements. Aerosol Sci. Technol., 42:636–657.
- Lack, D. A., Richardson, M. S., Law, D., Langridge, J. M., Cappa, C. D., Mclaughlin, R. J., and Murphy, D. M. (2012). Aircraft Instrument for Comprehensive Characterization of Aerosol Optical Properties, Part 2: Black and Brown Carbon Absorption and Absorption Enhancement Measured With Photo Acoustic Spectroscopy. Aerosol Sci. Technol., 46:555–568.
- Lamsal, L. N., Martin, R. V., von Donekelaar, A., Steinbacher, M., Celarier, E. A., Bucsela, E., Dunlea, E. J., and Pinto, J. P. (2008). Ground-Level Nitrogen Dioxide Concentrations Inferred From the Satellite-Borne Ozone Monitoring Instrument. J. Geophys. Res., 113:D16308, doi:10.1029/2007JD009235
- Lewis, K., Arnott, W. P, Moosmüller, H., and Wold, C. E. (2008). Strong Spectral Variation of Biomass Smoke Light Absorption and Single Scattering Albedo Observed With a Novel Dual-Wavelength Photoacoustic Instrument. J. Geophys. Res., 113:D16203, doi:10.1029/2007jd009699
- Li, Z., Xia, X., Cribb, M., Mi, W., Holben, B., Wang, P., Chen, H., Tsay, S.-C., Eck, T., Zhao, F., Dutton, E., and Dickerson, R. (2007). Aerosol Optical Properties and Their Radiative Effects in Northern China. J. Geophys. Res., 112:D22S01, doi:10.1029/2006JD007382.
- Li, J., Parchatka, U., Königstedt, R., and Fischer, H. (2012). Real-Time Measurements of Atmospheric CO Using a Continuous-Wave Room Temperature Quantum Cascade Laser Based Spectrometer. Opt. Exp., 20:7590–7601.
- Liu, S., Aiken, A. C., Gorkowski, K., Dubey, M. K., Cappa, C. D., Williams, L. R., Herndon, S. C., Massoli, P., Fortner, E. C., Chhabra, P. S., Brooks, W. A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., China, S., Sharma, N., Mazzoleni, C., Xu, L., Ng, N. L., Liu, D., Allan, J. D., Lee, J. D., Fleming, Z. L., Mohr, C., Zotter, P., Szidat, S., and Prévôt, A. S. H. (2015). Enhanced Light Absorption by Mixed Source Black and Brown Carbon Particles in UK Winter. Nat. Commun., 6:8435 doi:10.1038/ncomms9435
- Lobo, P., Durdina, L., Smallwood, G. J., Rindlisbacher, T., Siegerist, F., Black, E. A., Yu, Z., Mensah, A. A., Hagen, D. E., Miake-Lye, R. C., Thomson, K. A., Brem, B. R., Corbin, J. C., Abegglen, M., Sierau, B., Whitefield, P. D., and Wang, J. (2015). Measurement of Aircraft Engine Non-volatile PM Emissions: Results of the Aviation – Particle Regulatory Instrument Demonstration Experiment (A-PRIDE) 4 Campaign. Aerosol Sci. Technol., 49:472–484.
- Miklos, A., Hess, P, and Bozoki, Z. (2001). Application of Acoustic Resonators in Photoacoustic Trace Gas Analysis and Metrology. Rev. Sci. Instrum., 72:1937–1955.
- Molina, L. T., and Molina, M. J. (1986). Absolute Absorption Cross Sections of Ozone in the 185- to 350-nm Wavelength Range. J. Geophys. Res. D, 91:14501–14508.
- Montilla, E., Mogo, S., Cachorro, V., Lopez, J., and de Frutos, A. (2011). Absorption, Scattering and Single Scattering Albedo of Aerosols Obtained From in Situ Measurements in the Subarctic Coastal Region of Noway. Atmos. Chem. Phys. Discuss., 11:2162–2182.
- Moosmüller, H., Chakrabarty, R. K., and Arnott, W. P. (2009). Aerosol Light Absorption and Their Measurement: A Review. J. Quant. Spectrsc. Radiat. Transfer, 110:844–878.
- Mulholland, G. W., and Choi, M. Y. (1998). Measurement of the Mass Specific Extinction Coefficient for Acetylene and Ethene Smoke Using the Large Agglomerate Optics Facility. Proceedings of the 27th Symposium (International) on Combustion, vol. 1, August 2–7, Boulder, CO. Combustion Institute, Pittsburgh, PA, pp. 1515–1522.
- Nakayama, T., Suzuki, H., Kagaminani, S., Ikeda, Y., Uchiyama, A., and Matsumi, Y. (2015). Characterization of a Three Wavelength Photoacoustic Soot Spectrometer (PASS-3) and a Photoacoustic Extinctiometer (PAX). J. Meteor. Soc. Japan, 93:285–308.
- Osthoff, H. D., Brown, S. S., Ryerson, T. B., Fortin, T. J., Lerner, B. M., Williams, E. J., Pettersson, A., Baynard, T., Dubé, W. P., Ciciora, S. J., and Ravishankara, A. R. (2006). Measurement of Atmospheric NO2 by Pulsed Cavity Ring-Down Spectroscopy. J. Geophys. Res., 111:D12305, doi:10.1029/2005JD006942
- Petzold, A. and Schonlinner, M. (2004). Multi-Angle Absorption Photometer – A New Method for the Measurement of Aerosol Light Absorption and Atmospheric Black Carbon. J. Aerosol Sci., 35:421–441.
- Redmond, E. H., Dial, D. K., and Thompson, E. J. (2010). Light Scattering and Absorption by Wind Blown Dust: Theory, Measurement, and Recent Data. Aeolian Res., 2:5–26.
- Rosengren, L.-G. (1975). Optimal Optoacoustic Detector Design. Appl. Opt., 14:1960–1976.
- Rothman, L. S., et al. (2005). The HITRAN 2004 Molecular Spectroscopic Database. J. Quant. Spectrosc. Radiat. Transfer, 96:139–204.
- Russell, P. B., Bergstrom, R. W., Shinozuka, Y., Clarke, A. D., DeCarlo, P. F., Jimenez, J. L., Livingston, J. M., Redemann, J., Dubovik, O., and Strawa, A. (2010). Absorption Angstrom Exponent in AERONET and Related Data as an Indicator of Aerosol Composition. Atmos. Chem. Phys., 10:1155–1169.
- Scherer, J. J., Paul, J. B., O'Keefe, A., and Saykally, R. J. (1997). Cavity Ringdown Laser Absorption Spectroscopy: History, Development and Application to Pulsed Molecular Beams. Chem. Rev., 97:25–51.
- Sharma, N., Arnold, I. J., Moosmüller, H., Arnott, W. P., and Mazzoleni, C. (2013). Photoacoustic and Nephelometric Spectroscopy of Aerosol Optical Properties with a Supercontinuum Light Source. Atmos. Meas. Technol., 6:3501–3513.
- Stier, P., Seinfeld, H. J., Kinne, S., and Boucher, O. (2007). Aerosol Absorption and Radiative Forcing. Atmos. Chem. Phys., 7:5237–5261.
- Tian, G., Moosmüller, H., and Arnott, W. P. (2009). Simultaneous Photoacoustic Spectroscopy of Aerosol and Oxygen a-Band Absorption for the Calibration of Aerosol Light Absorption Measurements. Aerosol Sci. Technol., 43:1084–1090.
- Vandaele, A. C., Hermans, C., Fally, S., Carleer, M., Colin, R., Mériemme, M.-F., Jenouvrier, A., and Coquart, B. (2002). High-Resolution Fourier Transform Measurement of the NO2 Visible and Near-Infrared Absorption Cross Sections: Temperature and Pressure Effects. J. Geophys. Res., 107(D18):4348, doi:10.1029/2001JD000971
- Voigt, S., Orphal, J., and Burrows, J. P. (2002). The Temperature and Pressure Dependence of the Absorption Cross-Sections of NO2 in the 250–800 nm Region Measured by Fourier-Transform Spectroscopy. J. Photochem. Photobiol. A, 149:1–7.
- Weingartner, E., Saathoff, H., Schnaiter, M., Streit, N., Bitnar, B., and Baltensperger, U. (2003). Absorption of Light by Soot Particles: Determination of the Absorption Coefficient by Means of Aethalometers. Aerosol Sci., 34:1445–1463.
- Werle, P., Miicke, R., and Slemer, F. (1993). The Limits of Signal Averaging in Atmospheric Trace-Gas Monitoring by Tunable Diode-Laser Absorption Spectroscopy (TDLAS). Appl. Phys. B, 57:131–139.
- West, G. A., Barret, J. J., Siebert, D. R., and Reddy, K.V. (1983). Photoacoustic Spectroscopy. Rev. Sci. Instrum., 54:797–817.
- Xue, J., Li, Y., Wang, X., Durbin, T. D., Johnson, K. C., Karavalakis, G., Asa-Awuku, A., Villela, M., Quiros, D., Hu, S., Huai, T., Ayala, A., and Jung, H. S. (2015). Comparison of Vehicle Exhaust Particle Size Distributions Measured by SMPS and EEPS during Steady-State Conditions. Aerosol Sci. Technol., 49:984–996.