
Abstract
Highly oxidized multifunctional compounds (HOMs) formed through gas-phase reactions are thought to account for a significant fraction of the secondary organic aerosol (SOA) formed in low-nitric oxide (NO) environments. HOMs are known to be peroxide-rich and unstable in SOA, however, and their fate once they partition into particles is not well understood. In the study reported here, we identified particle-phase reactions and decomposition products for an α-alkoxy hydroperoxyaldehyde that served as a convenient model for HOMs, and also quantified rate and equilibrium constants for cyclic peroxyhemiacetal formation and the effects of particle acidity and relative humidity on reaction products and timescales for decomposition of peroxide-containing compounds. Sulfuric acid increased the rate of acetal formation and subsequent peroxide decomposition, but the effect was eliminated when aqueous seed particles were used in humid air, indicating that organic/aqueous phase separation can affect the ability of strong acids to catalyze these and other reactions in SOA. The results will be useful for understanding and predicting the atmospheric fate of organic peroxides and the effects of their particle-phase reactions on SOA composition.
Copyright © 2018 American Association for Aerosol Research
EDITOR:
Introduction
Highly oxidized multifunctional compounds (HOMs) formed by the reaction of ozone with α-pinene and by the oxidation of other volatile organic compounds (VOCs) have been measured as gas-phase (Ehn et al. Citation2012; Krapf et al. Citation2016) and particle-phase (Mutzel et al. Citation2015; Kristensen et al. Citation2016) reaction products in the laboratory and in the atmosphere. These HOMs have low volatility and are estimated to contribute over 50% of secondary organic aerosol (SOA) mass formed under pristine conditions (Ehn et al. Citation2014), and are expected to become a larger fraction of future SOA mass in urban areas as NOx concentrations decrease due to air quality regulations (Praske et al. Citation2018). HOMs are believed to be labile multifunctional hydroperoxides formed through gas-phase autoxidation reactions (Crounse et al. Citation2013) that allow a single VOC-oxidant reaction to lead to multiple additions of oxygen to organic molecules. A simple example of the formation of a hydroperoxycarbonyl through an autoxidation mechanism is shown in (Crounse et al. Citation2013), where the initial alkylperoxy radical (RO2•) is a key intermediate formed in the oxidation of nearly all VOCs (Orlando and Tyndall Citation2012). Upon condensing into particles, HOMs can decompose on timescales of tens of minutes or more (Mertes et al. Citation2012; Krapf et al. Citation2016), but due to the chemical complexity of HOMs and the SOA matrix, the particle-phase mechanisms by which they react have not been studied in detail. For example, mass spectrometric analysis of gas-phase HOMs produced from the reaction of α-pinene with O3 detected dozens of unique HOM molecular formulas corresponding to monomers and dimers with 5–20 carbons and 4–18 oxygens in each molecule (Ehn et al. Citation2012), and similar studies of SOA formed from this reaction have identified more than a thousand molecular formulas (Gao, Hall, and Johnston Citation2010). Since these studies rely on molecular formulas to assign products, they understate the chemical complexity of the HOMs and SOA by not being able to differentiate isomers or molecular structures. Studies of isoprene oxidation products have also observed that changes in aerosol acidity and phase state impact the condensed-phase chemistry of multifunctional peroxides (Riva et al. Citation2017; Zhang et al. Citation2018). For example, the reactive uptake of multifunctional hydroperoxides onto acidified seed aerosol can be suppressed by reduction of aerosol water in the seed particles and by organic coatings (Riva et al. Citation2017). Without greater knowledge of the molecular structures of the HOMs produced in these SOA systems, detailed studies of the particle-phase chemistry of HOMs are not feasible.
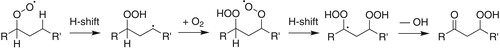
Figure 1. Simple representation of the autoxidation mechanism that leads to HOM formation from an alkylperoxy radical (Crounse et al. Citation2013). The resulting HOM in this scheme is a hydroperoxycarbonyl, like the AHPA model compound studied here.
In an attempt to overcome this difficulty, we recently investigated the kinetics, equilibria, products, and mechanisms of the particle-phase and solution reactions of 10-n-propoxy-10-hydroperoxydecanal [HOOCH(OCH2CH2CH3)(CH2)8CHO], a C13 α-alkoxy hydroperoxyaldehyde (AHPA) that is an appropriate model for HOMs. This compound was chosen because in addition to being hydroperoxide-rich, HOM monomers in SOA produced by α-pinene ozonolysis have been reported to contain 1–2 carbonyl groups per molecule (Mutzel et al. Citation2015; Krapf et al. Citation2016). The highly reactive hydroperoxide and carbonyl groups in these HOMs are expected to play a critical role in their particle-phase chemistry (Ziemann and Atkinson Citation2012), so it was necessary that the model compound employed here contain both structural features. A recent study also suggested that production rates of AHPA in the atmosphere can be significant under certain conditions (McGillen et al. Citation2017). Additional advantages of studying this AHPA are that it can be easily synthesized with high yields either in solution or in an environmental chamber as a major component of SOA, thus allowing for detailed studies on a single hydroperoxycarbonyl compound under well-characterized conditions, and that it has sufficiently low vapor pressure that its chemistry involves only particle-phase reactions (Ziemann Citation2003). The results of this study provide information that will be useful for understanding and predicting the atmospheric fate of HOMs and their contributions to SOA formation, lifetime, and composition.
Experimental
Chemicals
Chemicals with purities/grades and suppliers were as follows: trans-cyclodecene (98%), 1-propanol (99.5%) (Sigma-Aldrich, St Louis, MO, USA); tridecanoic acid (98%) (Aldrich, St Louis, MO, USA); 3-hexadecanone (99%) (Alfa Aesar, Haverhill, MA, USA); ethyl acetate (ACS) (Millipore, Burlington, MA, USA); bis-diethylhexyl sebacate (DOS) (97%) (Fluka, St Louis, MO, USA); ammonium sulfate (99.5%) (Fisher Scientific, Waltham, MA, USA); sulfuric acid (ACS) (Macron, Center Valley, PA, USA); nitric oxide (>99%) (Matheson Gas, Basking Ridge, NJ, USA); oxygen (UHP), carbon monoxide (CP) (AirGas, Radnor, PA, USA). Ozone was produced from oxygen using a LG7 ozone generator (Del Ozone, San Luis Obispo, CA, USA). The reagents and solvents used in functional group analysis have been described previously (Aimanant and Ziemann Citation2013a).
AHPA solution synthesis
The AHPA standards were prepared by dissolving 10 µL of cyclodecene in 10 mL of 1-propanol [CH3CH2 CH2OH] and bubbling 2% O3 through the solution at 1 L min−1 for 10 s to react with all the alkene. The mechanism of the reaction is shown in and discussed in the following section, but in solution produces AHPA with yields of ≥95% (Zelikman et al. Citation1971). Due to the potentially explosive nature of organic peroxides, researchers should take care when using this procedure. Total amounts of concentrated hydroperoxide should be kept to a minimum, especially when working with smaller peroxides than those synthesized here.
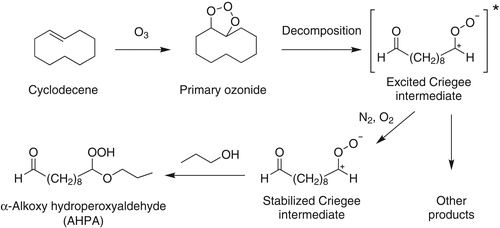
Figure 2. Mechanism of reaction of cyclodecene with O3 in the presence of 1-propanol to form 10-n-propoxy-10-hydroperoxy decanal (AHPA).
Environmental chamber reactions
AHPA was also formed as a component of SOA in an 8.0 m3 FEP Teflon environmental chamber by the gas-phase reaction of cyclodecene with O3 in the presence of 1-propanol. The chamber is operated at room temperature (∼25 °C) and pressure (∼630 Torr) and is flushed and filled with pure dry air (<5 ppb hydrocarbons, <1% RH) from an AADCO clean air generator. Reactions were typically conducted by adding seed particles, 1 ppm of cyclodecene, 1,500 ppm of 1-propanol, and then 30 ppm of O3 to initiate the reaction. Aerosol growth was complete within ∼5 min. After 10 min of reaction, which was sufficient to react with >95% of the cyclodecene (indicated by the cyclodecene decay measured with a proton transfer reaction-mass spectrometer (de Gouw and Warneke Citation2007) and estimated from the ∼1 min lifetime calculated from the rate constant of 2.9 × 10−17 cm3 molec−1 s−1 for cis-cyclodecene (Atkinson and Arey Citation2003) and the 30 ppm concentration of O3), 60 ppm of NO was added to remove the remaining O3 (O3 + NO → NO2 + O2). Also, in one experiment, particle-phase exchange reactions were probed by adding ∼4,000 ppm of methanol from 15 to 30 min after the aerosol stopped growing, and then adding ∼6,000 ppm of water (∼18% RH) 30–60 min later. And in another experiment, 630 ppm of carbon monoxide was used in place of 1-propanol as an SCI scavenger (>95% efficiency), with 50 ppm of cyclohexane used as an OH radical scavenger (Tobias and Ziemann Citation2001; Docherty and Ziemann Citation2003). All reactants were added by flushing measured amounts from a glass bulb (with or without heating) into the chamber using ultra-high purity (UHP) N2. Seed particles were either dioctyl sebacate (DOS), DOS/sulfuric acid, or aqueous ammonium sulfate/sulfuric acid. DOS was chosen because it is an inert, liquid organic compound with a sufficiently low vapor pressure to be essentially nonvolatile, for which we have developed methods for forming a nearly monodisperse seed aerosol. Use of DOS seed aerosol also allows for monitoring of particle wall losses using real-time mass spectrometry and the detection of SOA evaporation during a chamber reaction (Aimanant and Ziemann Citation2013b). The DOS particles were formed using an evaporation-condensation generator, and the DOS/sulfuric acid particles were formed by evaporating 0.25 mL of 0.1% sulfuric acid/water from a glass bulb and allowing the small sulfuric acid particles generated following the addition to coagulate with DOS seed particles produced by an evaporation-condensation generator. The coagulation was monitored with a scanning mobility particle sizer (SMPS) and the presence of sulfuric acid in the aerosol was confirmed by thermal desorption particle beam mass spectrometric (TDPBMS) analysis. These instruments are described in the following paragraphs. The aqueous ammonium sulfate/sulfuric acid particles were formed by atomizing an aqueous solution of 8 × 10−3 M ammonium sulfate and 8 × 10−5 M sulfuric acid from a Collison atomizer into the chamber at 50% RH. According to E-AIM calculations (www.aim.env.uea.ac.uk/aim/aim.php; Clegg, Brimblecombe, and Wexler Citation1998; Wexler and Clegg Citation2002), this aerosol had pH ∼1 once it equilibrated with the water vapor in the chamber. Throughout each experiment, the aerosol size distribution was measured using a scanning mobility particle sizer (SMPS) (Docherty and Ziemann Citation2003), aerosol composition was analyzed with a thermal desorption particle beam mass spectrometer (Tobias et al. Citation2000), and O3 was monitored with a Model 1003 ozone monitor (Dasibi, Glendale, CA, USA). After each experiment, aerosol was collected onto filters for offline analyses.
Mass spectrometric analysis
Aerosol is sampled directly into the thermal desorption particle beam mass spectrometer (TDPBMS) (Tobias et al. Citation2000) through an aerodynamic lens, which focuses the particles into a narrow beam for transport into a differentially pumped high-vacuum chamber. There they impact and vaporize on a polymer-coated copper rod that is heated to 160 °C, and the vapors are ionized by 70 eV electrons for analysis in an Extrel triple quadrupole mass spectrometer. To make explicit that TDPBMS mass spectra are obtained by electron ionization, hereafter we refer to this as the EI-TDPBMS.
The EI-TDPBMS response to AHPA aerosol was calibrated by generating known mass concentrations of aerosol containing synthesized AHPA. A 6.3 mM solution of the AHPA standard in 1-propanol (synthesized as described above) was atomized in a Collison atomizer that was coupled to a Nexus 3000 syringe pump (Chemyx, Stafford, TX, USA) and operated at 0.1 mL min−1. The aerosol produced was dried by passing through charcoal and silica gel diffusion dryers and then the flow was split, with 0.075 L min−1 entering the EI-TDPBMS and 5 L min−1 passing through a filter sampler for collection of particles onto pre-weighed PTFE filters (Millipore Fluoropore 0.45 μm) for 50 min. Filters were reweighed to determine the mass concentration of the AHPA aerosol entering the EI-TDPBMS and analyzed using peroxide functional group analysis to determine AHPA purity, which was >95%.
Solution-synthesized hydroperoxides and SOA collected on filter samples were also analyzed using a PolarisQ chemical ionization ion trap mass spectrometer (CIITMS) (Thermo Finnigan, Waltham, MA, USA) equipped with a direct insertion probe (Ranney and Ziemann Citation2017). Samples were prepared for CI-ITMS analysis by drying ∼3 μL of 5 mg mL−1 solution inside a small glass vial. The vials were then loaded into the tip of the direct insertion probe and thermally desorbed from the tip of the probe into the ionization region, where they underwent chemical ionization with isobutane as a reagent gas.
Filter sampling
Aerosol was collected onto pairs of pre-weighed PTFE filters (Millipore Fluoropore 0.45 μm) by sampling air at 15 L min−1 through two 30 cm long, ¼″ outer diameter stainless steel tubes. Filters were re-weighed after sampling and the mass of SOA collected was determined by difference. All mass measurements were conducted with a XS3DU microbalance (1 μg precision) (Mettler Toledo, Columbus, OH). The collected SOA was extracted into ethyl acetate without sonication (three 5-min extractions into 5 mL of ethyl acetate, which were then pooled) immediately following weighing to minimize the amount of time for SOA components to react while on the filter. Extraction efficiencies determined previously for these methods were >95% (Matsunaga and Ziemann Citation2009). When necessary, samples were stored overnight in a freezer at –25 °C. Peroxide functional group analysis was always conducted immediately after extraction (never frozen), and all other functional group analyses were conducted within 18 h of filter collection.
Functional group analysis
Derivatization-spectrophotometric methods we developed previously (Aimanant and Ziemann Citation2013a) were used to quantify the moles of peroxide [HC–OOH] (M = 46), carboxyl [O = C–OH] (M = 45), carbonyl [C = O] (M = 28), ester [O = C–O]–R (M = 44), hydroxyl [HC–OH] (M = 30), and methylene [CH2] (M = 14) functional groups in SOA, with the number of methylene groups being determined from the difference in the measured SOA mass and the mass of other functional groups calculated by assuming they have molecular weights corresponding to the structures shown in brackets. For convenience, it was assumed that all the products have 10 carbons, the same number as cyclodecene. This assumption is likely the largest source of error in our estimate of molecular weight, but since there was no evidence for the presence of oligomers in the SOA, the error should not be large. Each derivatization technique involves the formation of a chromophore specific to the functional group being measured, and solutions were then analyzed using an USB4000 UV-Vis spectrometer (Ocean Optics, Largo, FL, USA).
NMR analysis
Proton nuclear magnetic resonance spectroscopy (1H-NMR) analysis was carried out at the University of Colorado Department of Chemistry and Biochemistry NMR Spectroscopy Facility using a Avance-300 NMR spectrometer (Bruker, Billerica, MA, USA). AHPA standards were synthesized as described above and either analyzed as is or purified by collecting fractions manually from HPLC eluent. Fractions were dried under UHP N2 and reconstituted in deuterated dimethyl sulfoxide (d6-DMSO) for analysis. Spectra were processed using the MestReNova software package (Mestrelab Research, Santiago de Compostela, Spain). Without and with purification, the purity of AHPA determined from the NMR analysis was ≥95%.
Results and discussion
Mechanism of formation of AHPA
The mechanism of formation of the AHPA model HOM from reaction of cyclodecene with O3 in the presence of 1-propanol is shown in (Ziemann Citation2003). The reaction is initiated by addition of O3 to the C = C double bond to form an unstable primary ozonide, which rapidly decomposes to an excited Criegee intermediate (ECI). In solution, essentially all ECI are thermalized by collisions with solvent to form stabilized Criegee intermediates (SCI), which react almost exclusively with 1-propanol to form the AHPA, 10-n-propoxy-10-hydroperoxydecanal (Zelikman et al. Citation1971). In the gas phase, the ECI can be thermalized by collisions with air to form SCI, which can undergo ring closure to form a secondary ozonide (SOZ) (Donahue et al. Citation2011), or in the presence of a high concentration of 1-propanol, the SCI can react to form the AHPA. Using the approach presented by Docherty et al. (Citation2005), we estimate that the large excess of 1-propanol scavenges >95% of the stabilized Criegee intermediates and >99% of the OH radicals produced from the reaction. ECI also undergo isomerization and decomposition reactions that lead to stable products and the formation of RO2• radicals, which for the high VOC concentrations used here are expected to react with other RO2• radicals and HO2 radicals to form a variety of products containing carbonyl, hydroxyl, carboxyl, hydroperoxy, peroxycarboxyl (Ziemann Citation2002), and possibly ester (Müller et al. Citation2008) groups.
Yield of AHPA formed from the gas-phase reaction of cyclodecene with O3 in the presence of 1-propanol
In order to interpret the experimental results of this study, it was necessary to determine the molar yield of AHPA (same as the molar yield of SCI) formed from the gas-phase reaction of cyclodecene with O3 in the presence of 1-propanol. This was determined from the difference in the molar yields of peroxide in SOA (moles of peroxide in SOA/moles of cyclodecene reacted) formed when 1-propanol and CO were used as SCI scavengers, since reaction of CO with SCI forms CO2 and a volatile C10 dialdehyde (Atkinson Citation1997) rather than AHPA. From the measured molar yields of peroxide in SOA of 0.18 and 0.06 for the 1-propanol and CO experiments, we obtained a molar yield of AHPA of 0.12. For this reaction, we thus estimate that 12% of the products are formed through the SCI pathway and 88% through other pathways.
Potential reaction pathways of AHPA
A previous study of AHPA (Ziemann Citation2003) has shown that its hydroperoxide and carbonyl groups readily undergo an intramolecular reaction in SOA to form a cyclic peroxyhemiacetal (CPHA), as shown in , and that these are the dominant early reaction products. Intermolecular peroxyhemiacetal oligomer formation is also possible for AHPA, as it has been observed from the reaction of the C14 α-methoxy hydroperoxide and C13 aldehyde products of the ozonolysis of 1-tetradecene in the presence of methanol (Tobias and Ziemann Citation2000). In that study, the C27 peroxyhemiacetal was thermally desorbed intact for mass spectral analysis using temperature-programmed TDPBMS, and when the same technique was applied to AHPA SOA no C26 peroxyhemiacetal oligomer (which would have been the product formed from reaction of two C13 AHPA molecules) was detected, indicating that AHPA does not undergo intermolecular peroxyhemiacetal formation (Ziemann Citation2003). It is also worth noting that evidence for the stability of peroxides during thermal desorption under the conditions used here can be found in the EI-TDPBMS and CI-ITMS mass spectra shown in , which can only be explained by fragmentation of the [M]+• and [M + H]+ ions formed by ionization of the intact AHPA.
Figure 3. Potential reactions of AHPA in SOA. In the concerted elimination and strong acid decomposition pathways H2, water, and propanol co-products are not shown.
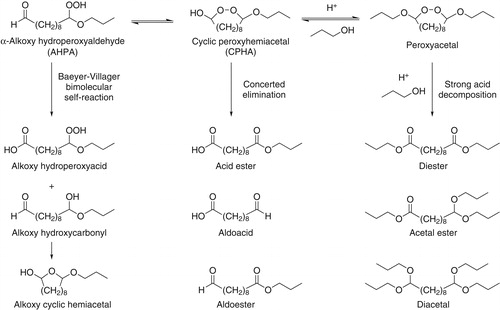
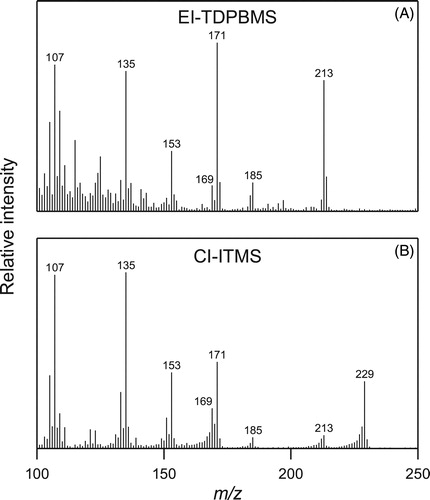
Figure 4. Mass spectra of AHPA standard obtained via EI-TDPBMS and CI-ITMS.
Since relatively little is known about the reactions of hydroperoxides in SOA beyond peroxyhemiacetal formation, and since CPHA have structures similar to the cyclic hemiacetals (CHA) formed in SOA from cyclization of 1,4-hydroxycarbonyls (Aimanant and Ziemann Citation2013b; Ranney and Ziemann Citation2016), we have looked into the chemistry of 1,4-hydroxycarbonyls and into the synthetic organic chemistry literature to develop a more complete scheme for describing the potential fate of AHPA.
After 1,4-hydroxycarbonyls cyclize to form CHA they can undergo dehydration to form dihydrofurans, or oligomerization in which CHA undergo self-reactions to form acetals (Aimanant and Ziemann Citation2013b). Studies conducted on CHA in dry SOA in the presence of added HNO3 showed that the dehydration of CHA is catalyzed by undissociated molecular HNO3 (general acid catalysis) (Ranney and Ziemann Citation2016), whereas acetal formation is apparently catalyzed by H+ (specific acid catalysis) (Ziemann and Atkinson Citation2012). On the basis of these studies, CPHA is expected to undergo either dehydration or reaction with 1-propanol to form a peroxyacetal. 1-Propanol is shown as the reacting alcohol in because its mixing ratio in the chamber (1,500 ppm to ensure high-efficiency scavenging of SCI) was so high that it provided a controlled source of hydroxyl groups for particle-phase reactions. Knowing the identity of the reacting alcohol in the SOA facilitated the identification of products of accretion reactions.
Two studies of the decomposition of organic hydroperoxides in the presence of aldehydes (Durham, Wurster, and Mosher Citation1958) and strong acid (Griesbaum and Neumeister Citation1982) reported in the synthetic organic chemistry literature are also relevant to the current work, even though they were conducted in bulk solutions instead of SOA. In particular, they are informative since they establish that AHPA cyclize to CPHA, and they point to the importance of peroxyhemiacetals as key intermediates in decomposition reactions. In their study of the decomposition of n-butyl hydroperoxide in the presence of butanal, Durham, Wurster and Mosher (Citation1958) observed a high yield (0.52) of gaseous H2, and experiments with deuterated peroxide showed that the source of H2 was the alkyl chains of the hydroperoxide and aldehyde. The primary products in solution were butyric acid, butyraldehyde, butyl butyrate, butyl alcohol, and a small amount of butyl formate, indicating that decomposition did not proceed by a single pathway under these conditions. And when Griesbaum and Neumeister (Citation1982) investigated the decomposition of 6-methoxy-6-hydroperoxyhexanal (an AHPA similar to ours) in methanol in the presence of strong acid, they observed a 1:2:1 yield ratio of diester, acetal ester, and diacetal, and proposed a simpler decomposition mechanism in which a peroxyacetal intermediate decomposed to give these products. These results have been incorporated into as the concerted elimination and strong acid decomposition pathways for CHPA, respectively. Knowing then that CPHA and peroxyacetals are likely to be key intermediates in AHPA decomposition in SOA, we set out to characterize the cyclization reaction of AHPA and identify decomposition products with and without strong acid present to determine which of these pathways are likely applicable to HOMs.
Mass spectra of AHPA standards
Mass spectra of the AHPA (M = 246) standard acquired using EI-TDPBMS and CI-ITMS are shown in . The electron ionization fragmentation pattern has been established previously (Ziemann Citation2003), with dominant peaks at m/z 213 and 171. Under chemical ionization conditions, the AHPA shows a similar fragmentation pattern but with an additional peak at m/z 229. These fragmentation pathways are described further in .
Table 1. Characteristic ions and fragmentation pathways for major products in SOA analyzed by EI-TDPBMS and CI-ITMS.
Rate and equilibrium constants for AHPA cyclization
Rate and equilibrium constants for cyclization of hydroperoxyaldehydes have not been previously reported, and SOA models (Capouet et al. Citation2008) have only included peroxyhemiacetal formation from bimolecular reactions using values obtained from measurements conducted in bulk solutions (Antonovskii and Terent’ev Citation1967). In an effort to gain more quantitative information on this reaction, the formation of AHPA and cyclization to CPHA in SOA produced in the environmental chamber from the reaction of cyclodecene with O3 in the presence of 1-propanol and DOS seed particles were monitored using the EI-TDPBMS signals at m/z 169 and 171, which are characteristic ions formed by electron ionization of CPHA and AHPA, respectively (Ziemann Citation2003). A plot of the ratio of these signals is shown in , with the ratio reaching an equilibrium plateau at ∼10 min. An exponential fit to this profile yields a timescale to reach equilibrium of τeq = 2.7 min. In addition, an equilibrium constant of Keq = 2.6 was calculated for the reaction from EquationEquation (1)(1) :
(1)
where, [AHPA]eq, [CPHA]eq, and [AHPA + CPHA]eq are the concentrations of AHPA, CPHA, and AHPA + CPHA in the SOA at equilibrium. [AHPA]eq was determined from the m/z 171 signal at the plateau using the calibration procedure described above, and [AHPA + CPHA]eq was determined from offline peroxide analysis and the ratio of the AHPA molar yield/total peroxide molar yield of 0.12/0.18 reported above using EquationEquation (2)
(2) :
(2)
Figure 5. Time profile of the ratio of real-time EI-TDPBMS mass spectral signals measured at peaks characteristic of CPHA (m/z 169) and AHPA (m/z 171) in SOA formed from reaction of cyclodecene with O3 in the presence of 1-propanol and DOS seed particles. The data were fit to an exponential function to determine the timescale to establish cyclization equilibrium (τeq) and the equilibrium constant (Keq).
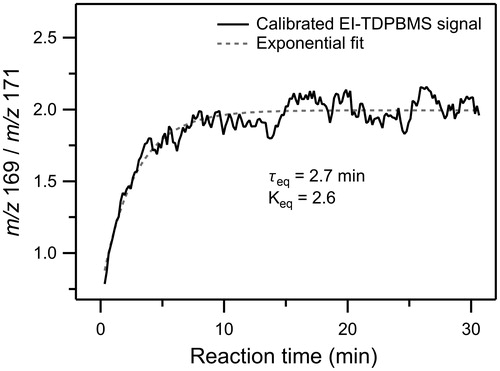
We also measured a similar equilibrium constant of 2.8 for the cyclization of AHPA in d6-DMSO using 1H-NMR, with peak assignments and integrals presented in Table S1 in the online supplementary information (SI). The rate constants for the cyclization (kf) and ring-opening reactions (kr) were then calculated from τeq = 2.7 min and Keq = 2.6 to be kf = 4.4 × 10−3 s−1 and kr = 1.7 × 10−3 s−1 using EquationEquations (3)(3) and Equation(4)
(4) (Schwarzenbach, Gschwend, and Imboden Citation2003):
(3)
(4)
This measured timescale of ∼3 min for cyclization can be compared with values expected for particle-phase bimolecular reactions. From experiments conducted with tert-butyl hydroperoxide (a tertiary hydroperoxide with a structure most similar to the secondary hydroperoxide used here) and acetaldehyde in different solvents, Antonovskii and Terent’ev (Citation1967) report a range of room temperature rate constants of 1.2 × 10−2 to 1.3 × 10−4 M−1 s−1 for bimolecular peroxyhemiacetal formation and 2.0 × 10−4 to 8.3 × 10−5 s−1 for the reverse reaction.
These rate constants were used in kinetic simulations conducted in KinSim v3.1 (Peng et al. Citation2015) with the following quantities to give a range of timescales for establishing equilibrium of about 6 to 80 min: initial concentrations of hydroperoxide and aldehyde groups of 2 M (estimated assuming an SOA density of 1.2 g cm−3), an average SOA molecular weight of 250 g mol−1 (calculated from the results of the functional group analysis [Aimanant and Ziemann Citation2013a]), and a mole fraction of AHPA in the SOA of 0.43 (calculated from the product of the molar yields of AHPA (0.12) and SOA (0.36), with the latter value calculated as (mass of SOA/molecular weight of SOA)/moles of cyclodecene reacted). Cyclization is faster than all of these reactions, probably because unimolecular reactions do not have the same unfavorable loss of entropy as bimolecular reactions (Bruckner Citation2002). The rates of both cyclization and oligomer formation could be increased by the presence of a strong acid catalyst, but this also affects rates of decomposition reactions as discussed below.
Peroxide decomposition
Prior to investigating the decomposition of peroxides present in SOA, the possible effects of filter sampling, extraction, and weighing on their stability was determined by atomizing synthesized AHPA (purity ≥95%) onto a PTFE filter, extracting the AHPA in ethyl acetate, and measuring the mass and peroxide content of the extract. Comparison of the extracted moles of AHPA based on measured SOA mass (moles AHPA = SOA mass/AHPA molecular weight) and measured peroxide (moles AHPA = moles peroxide) showed that the sample was still ≥95% AHPA, indicating that filter collection and extraction did not contribute to the peroxide decomposition reported here for SOA. In addition, filter mass was stable over the 3 min required to take replicate measurements on the microbalance.
The rate of decomposition of AHPA (and other peroxides present in smaller amounts) was then measured for SOA formed under the following three conditions: DOS seed particles at ∼0% RH, DOS/sulfuric acid seed particles at ∼0% RH, and aqueous ammonium sulfate/sulfuric acid seed particles at pH ∼1 and 50% RH, which is typical of atmospheric conditions (Guo et al. Citation2015, Citation2017). After SOA formation, 3–6 filter samples were collected at different intervals over a period of ∼4 h, extracted, and analyzed immediately for peroxide content. We note that no nucleation was observed in any experiment with seed aerosol, and that functional group analysis of experiments without seed aerosol gave the same results as those using dry DOS seed. Furthermore, the SOA mass concentration in the chamber was constant after correcting for particle wall loss, indicating that no significant particle evaporation occurred during the experiments.
The results of the peroxide decomposition measurements are shown in , where the number of moles of SOA was calculated from the SOA mass and the average SOA molecular weight of 250 g mol−1. Under dry conditions with inert DOS seed particles, the decomposition rate was 17% h−1. This is similar to the rates of 13% h−1 and 15% h−1 measured by Mertes et al. (Citation2012) and Krapf et al. (Citation2016) for SOA formed from α-pinene ozonolysis. Despite the differences in peroxide structure and SOA composition, the emerging trend is that organic peroxides in SOA decompose within a few hours when there are no strong acids present. In experiments conducted with seed particles containing sulfuric acid, the effect of the sulfuric acid on peroxide decomposition varied with the type of particles used. In experiments using dry DOS/sulfuric acid seed particles, the peroxide content of the SOA had fallen to almost zero by the time a filter sample could be collected after 30 min of reaction (), indicating a timescale less than ∼10 min. When aqueous ammonium sulfate/sulfuric acid seed particles were used, however, the peroxide decomposition rate was similar to that for dry DOS seed particles without added acid.
Figure 6. Time profiles of peroxide functional group content of SOA formed from the reaction of cyclodecene with O3 in the presence of 1-propanol and seed particles with the following composition: (squares) aqueous ammonium sulfate/sulfuric acid at 50% RH and pH = 1, (circles) dry DOS, and (diamonds) dry DOS/sulfuric acid.
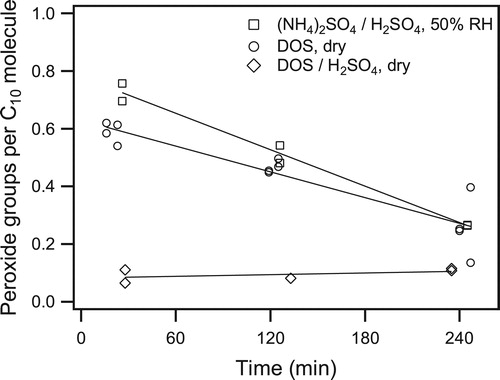
We attribute the difference in decomposition observed with dry DOS/sulfuric acid and aqueous ammonium sulfate/sulfuric acid seed particles to the phase state of the particles and its effect on partitioning of sulfuric acid. The parameterization of Bertram et al. (Citation2011) shows that SOA with an O/C ratio of 0.3 (calculated from the functional group composition) and an ammonium sulfate component will exhibit liquid-liquid phase separation between the organic and aqueous phases below 95% RH. The experiments in this work using aqueous ammonium sulfate/sulfuric acid seed particles were conducted at 50% RH, so we expect two phases to be present when AHPA SOA condenses onto the seed particles. Although the morphology of these phase-separated particles is unknown, for organic aerosol with an average molecular weight of 250 g mol−1 and no oligomers, the estimated timescale for a molecule to diffuse through a particle of the size generated in these experiments is ∼1 ms (Docherty and Ziemann Citation2006). Since this timescale is much shorter than the observed timescale for peroxide decomposition (∼6 h), the organic phase is likely well-mixed during peroxide decomposition regardless of morphology. Furthermore, although published research investigating the partitioning of sulfuric acid between aqueous and organic phases is sparse, one reported study (Hayes and Pepper Citation1961) conducted with dichloroethane indicates that only a trace amount of undissociated molecular sulfuric acid partitions to the organic phase. This is consistent with our E-AIM (Wexler and Clegg Citation2002) calculations that show 100% of the sulfuric acid is dissociated in the aqueous phase, making molecular sulfuric acid unavailable for partitioning to the organic SOA phase. Together these results suggest that sulfuric acid exists almost entirely in the aqueous phase in these experiments and so is probably not available to catalyze AHPA decomposition in the organic phase.
These results highlight the uncertain nature of acid catalysis in aerosol where there is little water or there is phase separation between organic and aqueous phases. Recent results showing that the pH range of ambient submicron aerosols is typically 0–4 assume that hygroscopic organic components are in the aqueous phase and contribute to total aerosol water uptake (Guo et al. Citation2015, Citation2017). Our results indicate, however, that for phase-separated aerosol with low O/C ratios, such as AHPA, most of the acid remains in the aqueous phase and so is not available to catalyze reactions in the organic phase. Phase separation may thus create a potential barrier to acid catalysis in aerosol particles. In light of the results of Ranney and Ziemann (Citation2016) that demonstrated occurrence of general acid catalysis by undissociated molecular nitric acid in dry organic aerosol, it seems plausible that the acid-catalyzed decomposition that we observe in dry aerosol using organic seed is general acid catalysis by undissociated molecular sulfuric acid. The mechanisms of general acid catalysis in concentrated solutions are well documented, with prior studies of the acid-catalyzed hydration of alkenes and acid-catalyzed dehydration of alcohols showing that these reactions can proceed through a wide variety of general acid catalysis mechanisms, depending on the identity and concentration of the acid catalyst and the properties of the solvent (Vinnik and Obraztsov Citation1990). It seems likely, therefore, that acid catalysis in aerosols proceeds by different mechanisms in different aerosol phases, and that the mechanisms of reaction will vary with aerosol phase and humidity.
Identification of peroxide decomposition products
Using the EI-TDPBMS and CI-ITMS mass spectra, functional group composition, and potential reaction pathways outlined above as guides, we have identified several decomposition products of AHPA. The structure, molecular weight, characteristic ions, and EI and CI fragmentation pathways for all the major identified reaction products are shown in . shows real-time EI-TDPBMS spectra of the SOA formed in the absence and presence of sulfuric acid and humidity. shows the mass spectrum immediately following SOA formation on dry DOS seed particles in the absence of sulfuric acid and shows the mass spectrum of this SOA after 4 h of peroxide decomposition. Most notable is the similarity of the two mass spectra. Whereas we attribute the peaks at m/z 213 and 171 in to AHPA, and m/z 229 and 169 to CPHA (Ziemann Citation2003), their presence in must also be due to other compounds since we know from functional group analysis that ∼60% of the peroxides have decomposed within 4 h (). Products that are expected to have peaks at m/z 229, 213, 171, and 169 could be formed from a bimolecular Baeyer–Villager reaction (Wurster, Durham, and Mosher Citation1958) between two AHPA, which are present in significant amounts ([AHPA]eq/[CPHA]eq ∼ 0.4) after rapid equilibration with CPHA. In this reaction, a hydroperoxide group in one AHPA oxidizes the carbonyl group of another AHPA to a carboxyl group and in the process is reduced to a hydroxyl group (). The resulting products are an alkoxy hydroperoxyacid and alkoxy hydroxycarbonyl, with the latter compound rapidly cyclizing to an alkoxy cyclic hemiacetal (Ranney and Ziemann Citation2016). They should undergo EI fragmentation by pathways similar to those of AHPA and CHPA (Ziemann Citation2003) to form ions at m/z 229, 213, 171, and m/z 169. It is also likely that over the 4 h period of peroxide decomposition that the peroxyacetal is formed () in significant amounts and contributes to the m/z 229 and 169 peaks by fragmentation pathways similar to the CPHA.
Figure 7. Real-time EI-TDPBMS mass spectra measured at different times for SOA formed from the reaction of cyclodecene with O3 in the presence of 1-propanol and seed particles with different compositions as follows: (a) immediately after formation on dry DOS, (b) 4 h after formation on dry DOS, (c) immediately after formation on dry DOS/sulfuric acid, and (d) 4 h after formation on aqueous ammonium sulfate/sulfuric acid at 50% RH and pH = 1.
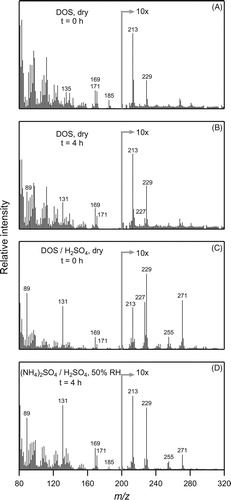
When real-time EI-TDPBMS mass spectra were taken as SOA peroxides decomposed (), peaks at m/z 131 and 89 increased with time regardless of the absence or presence of sulfuric acid or humidity. These peaks were observed as a pair in all instances, and are characteristic of α-cleavage of dipropoxy acetals (Friedel and Sharkey Citation1956; Borisova et al. Citation1981). Several reference spectra of dipropoxy acetals are also available in the NIST Chemistry WebBook and the AIST Spectral Database that display peaks at m/z 89 and 131, and these are shown in Figure S1 (Stein Citation2016; SDBSWeb Citation2017). The acetal ester and diacetal shown in (two of the three products of peroxyacetal decomposition), are the likely source of these peaks. Formation of the acetal ester, diacetal, and diester are consistent with the peaks at m/z 271, 255, and 227, respectively, in and (McLafferty Citation2012).
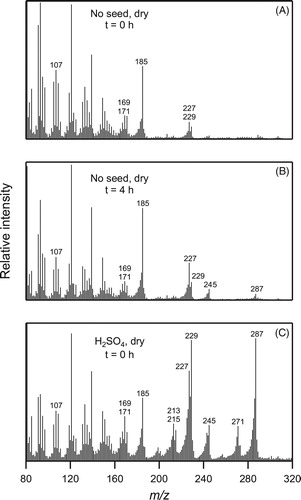
Further evidence of the formation of peroxyacetals and subsequent decomposition to the acetal ester and diester are seen in the CI-ITMS mass spectra in of SOA formed in the absence ( and ) and presence () of dry sulfuric acid seed particles. Looking first at , ionization of the acetal ester should create an [M + H]+ ion that fragments to form m/z 271 and 229 ions that are characteristic of acetals (Borisova et al. Citation1981; Lin, Tien, and Chang Citation1989). Evidence for the diester was obtained by comparing the mass spectrum with that of bis-diethylhexyl sebacate (DOS), a sebacate diester standard, for which we observed a [M + H]+ peak along with peaks corresponding to losses of an alkene, alcohol, and both simultaneously, from the alkyl substituents. The corresponding peaks for the diester are m/z 287, 245, 227, and 185, all of which are intense in the mass spectrum. Peaks from the acetal ester and (to a lesser extent) the diester are also present in mass spectra of the SOA formed and aged in the absence of sulfuric acid seed particles ( and ), though they are less intense due to the reduced decomposition of peroxides. In the mass spectra shown in and it is likely that the CPHA and peroxyacetal, which are precursors to acetal ester and diester formation, also contribute to the m/z 229 and 169 and peaks. The formation of the acetal ester, diester, and diacetal products is consistent with the mechanism proposed by Griesbaum and Neumeister (Citation1982), but only the real-time EI-TDPBMS mass spectrum provides plausible evidence for the formation of the diacetal that they propose. It is possible that peaks due to the diacetal are absent in the CI-ITMS mass spectrum because of ion suppression by the esters, which is a phenomenon we have observed in some analyses. The presence of the acid ester, aldoacid, and aldoester products of CPHA decomposition () cannot be verified from the mass spectra, since their dominant peaks in EI-TDPBMS and CI-ITMS analysis are likely to be m/z 185 and/or 169, which overlap with peaks assigned to fragmentation of other products that are more definitively identified by characteristic high-mass peaks.
Figure 8. CI-ITMS mass spectra of filter extracts of SOA collected at different times from the reaction of cyclodecene with O3 in the presence of 1-propanol in the absence or presence of seed particles as follows: (a) immediately after formation in the absence of seed particles, (b) 4 h after formation in the absence of seed particles, and (c) immediately after formation on dry sulfuric acid seed particles. DOS seed particles were not used in these experiments because DOS is the dominant signal in CI-ITMS at the concentrations used in chamber experiments.
The formation of the acetal ester, diester, and diacetal from decomposition of the peroxyacetal is also supported by the results of SOA functional group analysis presented in . By comparing the functional group composition of SOA produced in the presence of CO (which does not include products formed by the SCI pathway) to that produced in the presence of 1-propanol (which includes the same products as in the CO experiment plus AHPA formed by the SCI pathway) and sulfuric acid seed aerosol (which leads to rapid peroxide decomposition), one can infer the functionality of the products of peroxyacetal decomposition since they are formed from AHPA. Complications can occur in the interpretation of functional group analyses when significant amounts of peroxides decompose during an analysis, which is why we do not present results for experiments where significant amounts of AHPA are still present. For the two experiments presented in , that was not an issue since almost no peroxide was present in either SOA. The greater yield of ester (0.20 compared to 0.10) and carbonyl (0.47 compared to 0.20) groups in the SOA formed in the presence of 1-propanol and sulfuric acid is indicative of the formation of the acetal ester, diester, and diacetal, since during carbonyl analysis acetals are hydrolyzed to form carbonyl (and hydroxyl) groups, and these carbonyl groups are then derivatized and contribute to the measured carbonyl content. The higher SOA molar yield for that experiment (0.36 compared to 0.19), which is close to the sum of the yields (0.31) for the CO experiment (0.19) and AHPA (0.12), is also to be expected since AHPA is the sole product of the SCI reaction with 1-propanol and is the precursor to the peroxyacetal. The observed lower yield of carboxyl groups (0.14 compared to 0.18) is not as easily explained, but is possibly due to particle-phase reactions of oxoacids and hydroxyacids in the aerosol. These compounds comprise a significant fraction of SOA produced from cycloalkene ozonolysis (Gao et al. Citation2004), and could have undergone acid-catalyzed cyclization to form more volatile hydroxylactones and cyclic esters that evaporated from the aerosol and so lowered the yield of carboxyl groups. The consistency of the functional group analysis results (which are obtained without use of high temperatures) with the products of AHPA decomposition identified using EI-TDPBMS and CI-ITMS give us confidence that the high temperatures used to vaporize SOA in those instruments did not introduce significant artifacts in our results. It is also worth noting that the occurrence of peroxide decomposition in the presence of either DOS or DOS/sulfuric acid seed particles suggests that a strong acid is not necessary to catalyze this process. For example, from EI-TDPBMS mass spectra (Figure S2), we identified sebacic acid as a major component of the SOA produced by a non-SCI pathway, and it is possible that organic acids such as this are able to catalyze CHPA and peroxyacetal decomposition. The larger decrease in the amount of peroxide groups and larger increase in the amounts of carbonyl and ester groups and the peaks at m/z 89 and 131 in EI-TDPBMS mass spectra, in experiments when sulfuric acid seed was present, indicate that sulfuric acid is more efficient than organic acids at catalyzing peroxide decomposition.
Table 2. Molar yields of functional groups in SOA formed in the presence of CO without seed particles or 1-propanol with sulfuric acid seed particles.
Particle-phase alcohol and water exchange reactions
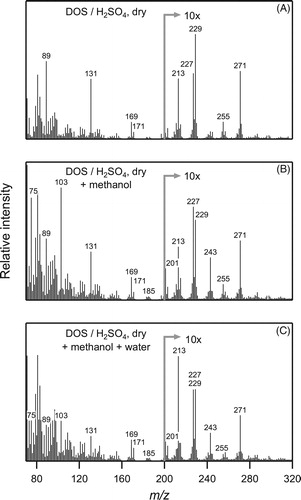
Since the decomposition products identified above are oligomers formed by reactions involving the most abundant alcohol in the SOA (1-propanol), understanding the reactions between CPHA and alcohols in SOA is central to understanding how HOMs will react with alcohols in atmospheric aerosol. To explore this question, an environmental chamber experiment was conducted to probe the exchange of alkoxy groups in SOA products due to competition with other alcohols or water. After forming SOA from the reaction of cyclodecene with O3 in the presence of 1-propanol and dry DOS/sulfuric acid seed particles, ∼4,000 ppm of methanol was added to the chamber, and then ∼6,000 ppm of water (18% RH). As shown in , upon addition of methanol peaks at m/z 103 and 75 grew in relative to that at m/z 131 due to exchange of one and then two propoxy groups for methoxy groups in the acetal ester and diacetal. High-mass peaks at m/z 243, 227, and 201 also grew in relative to those at m/z 271, 255, and 229 due to these exchange reactions, but since fragmentation of the acetal ester and diacetal occur by loss of alcohols, alkenes, and alkoxy radicals, changes in high-mass peaks did not provide information beyond the exchange of one methoxy and propoxy group. When water was added, peaks at m/z 131, 103, 89, and 75 all decreased significantly, due to exchange of methoxy and propoxy for hydroxyl groups. Although the relative intensities of some of the high-mass peaks also changed, no significant new peaks grew in. This is probably due to dissociation of the hemiacetal and gemdiol products to aldehydes, alcohols, and water that then evaporated from the particles because of their high volatility.
Figure 9. Real-time EI-TDPBMS mass spectra of (a) SOA formed from the reaction of cyclodecene with O3 in the presence of 1-propanol and dry DOS/sulfuric acid seed particles, and with the subsequent addition of (b) methanol, and then (c) water.
Conclusions
The results of this study provide new information on the potential influence of particle-phase chemistry on the atmospheric fate of hydroperoxycarbonyl compounds that can be formed under low-NO conditions, including HOMs that are formed through autoxidation reactions. Experiments conducted in an environmental chamber and in solution using a model α-alkoxy hydroperoxyaldehyde formed from the ozonolysis of cyclodecene in the presence of 1-propanol indicate that these compounds rapidly cyclize in particles to cyclic peroxyhemiacetals, reaching an ∼3:1 equilibrium mixture of the cyclic and acyclic forms in ∼3 min. Based on the results of SOA mass spectral and functional group analysis, we propose that a number of different reaction pathways then lead to cleavage of the O–O peroxide bond and formation of a variety of decomposition products. In particular, it appears that the α-alkoxy hydroperoxyaldehyde decomposes through a bimolecular Baeyer-Villager self-reaction and that the cyclic peroxyhemiacetal either decomposes by a unimolecular reaction or first reacts with alcohol (in this case 1-propanol) to form a peroxyacetal that then undergoes unimolecular decomposition. In SOA formed in the absence of sulfuric acid or in particles apparently containing separate SOA and aqueous ammonium sulfate/sulfuric acid phases decomposition occurred on a timescale of ∼6 h, similar to other reported studies, whereas in dry SOA containing sulfuric acid the timescale was at most ∼10 min. These timescales are all significantly shorter than the 6-day peroxide photolysis lifetime measured for SOA produced from α-pinene ozonolysis (Epstein, Blair, and Nizkorodov Citation2014), indicating that non-photolytic decomposition reactions of the type observed here are the dominant loss processes for hydroperoxycarbonyls in SOA. The fate of these compounds in the atmosphere should thus depend on the composition of the SOA, particle acidity, and relative humidity.
It is important to note here that because of differences in gas-phase reaction mechanisms, hydroperoxycarbonyls formed in the atmosphere would most likely contain an alkyl group instead of the alkoxy group that is present in the model α-alkoxy hydroperoxyaldehyde, cyclic peroxyhemiacetal, and their decomposition products. Despite this difference, the consistency of our results with prior studies of rates (Mertes et al. Citation2012; Krapf et al. Citation2016) and products of decomposition of peroxyhemiacetals (Durham, Wurster, and Mosher Citation1958) leads us to believe that the decomposition pathways of a hydroperoxycarbonyl with an alkoxy substituent are similar to those of a hydroperoxycarbonyl with an alkyl substituent. The alkoxy hydroperoxyacid, alkoxy hydroxycarbonyl, alkoxy cyclic hemiacetal, acid ester, aldoacid, and aldoester decomposition products () would then become a hydroperoxyacid, hydroxycarbonyl, cyclic hemiacetal, ketoacid, and dicarbonyl (Figure S3). And for the peroxyacetal and its decomposition products, one of the alkoxy groups would be replaced by an alkyl group so that the diester, acetal ester, and diacetal would then become a ketoester, ketoacetal, hydroxyester, and hydroxyacetal (Figure S3). Furthermore, in the atmosphere, the alkoxy groups in all these compounds would have been added by reactions with the large variety of low volatility multifunctional compounds containing hydroxyl groups (instead of 1-propanol) that are present in SOA, leading to the formation of much larger oligomeric compounds. These compounds would in turn be amenable to acid-catalyzed exchange reactions with other alcohols or water.
Supplemental Material
Download Zip (351.7 KB)Additional information
Funding
Related Research Data
References
- Aimanant, S., and P. J. Ziemann. 2013a. Development of Spectrophotometric Methods for the Analysis of Functional Groups in Oxidized Organic Aerosol. Aerosol Sci. Technol. 47 (6):581–91. doi: 10.1080/02786826.2013.773579.
- Aimanant, S., and P. J. Ziemann. 2013b. Chemical Mechanisms of Aging of Aerosol Formed from the Reaction of n-Pentadecane with OH Radicals in the Presence of NOx. Aerosol Sci. Technol. 47 (9):979–90. doi: 10.1080/02786826.2013.804621.
- Antonovskii, V. L., and V. A. Terent’ev. 1967. Effect of the Structure of Hydroperoxides and Some Aldehydes on the Kinetics of the Noncatalytic Formation of α-Hydroxy Peroxides. Zhurnal Org. Khimii. 3:1011–15.
- Atkinson, R. 1997. Gas-Phase Tropospheric Chemistry of Volatile Organic Compounds: 1. Alkanes and Alkenes. J. Phys. Chem. Ref. Data. 26 (2):215–90. doi:10.1063/1.556012.
- Atkinson, R., and J. Arey. 2003. Atmospheric Degradation of Volatile Organic Compounds. Chem. Rev. 103 (12):4605–38. doi:10.1021/cr0206420.
- Bertram, A. K., S. T. Martin, S. J. Hanna, M. L. Smith, A. Bodsworth, Q. Chen, M. Kuwata, A. Liu, Y. You, and S. R. Zorn. 2011. Predicting the Relative Humidities of Liquid-Liquid Phase Separation, Efflorescence, and Deliquescence of Mixed Particles of Ammonium Sulfate, Organic Material, and Water Using the Organic-to-Sulfate Mass Ratio of the Particle and the Oxygen-to-Carbon Elemental Ratio of the Organic Component. Atmos. Chem. Phys. 11 (21):10995–1006. doi:10.5194/acp-11-10995-2011.
- Borisova, I. A., V. I. Kadentsev, S. S. Zlotskii, O. S. Chizhov, M. R. Skurko, D. L. Rakhmankulov, and R. A. Karakhanov. 1981. Behavior of Acetals Under Chemical Ionization Conditions. Russ. Chem. Bull. 30 (7):1219–24. doi: 10.1007/BF01417976.
- Bruckner, R. 2002. Advanced Organic Chemistry. San Diego, CA: Harcourt/Academic Press.
- Capouet, M., J. F. Müller, K. Ceulemans, S. Compernolle, L. Vereecken, and J. Peeters. 2008. Modeling Aerosol Formation in Alpha-Pinene Photo-Oxidation Experiments. J. Geophys. Res. 113 (D2):D02308. doi:10.1029/2007JD008995.
- Clegg, S. L., P. Brimblecombe, and A. S. Wexler. 1998. A Thermodynamic Model of the System H+–NH4+–Na+–SO42−–NO3—Cl−–H2O at 298.15 K. J. Phys. Chem. A. 102 (12):2155–71. doi:10.1021/jp973043j.
- Crounse, J. D., L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, and P. O. Wennberg. 2013. Autoxidation of Organic Compounds in the Atmosphere. J. Phys. Chem. Lett. 4 (20):3513–20. doi:10.1021/jz4019207.
- de Gouw, J., and C. Warneke. 2007. Measurements of Volatile Organic Compounds in the Earth’s Atmosphere Using Proton-Transfer-Reaction Mass Spectrometry. Mass Spec. Rev. 26 (2):223–57. doi:10.1002/mas.20119.
- Docherty, K. S., W. Wu, Y. B. Lim, and P. J. Ziemann. 2005. Contributions of Organic Peroxides to Secondary Aerosol Formed from Reactions of Monoterpenes with O3. Environ. Sci. Technol. 39 (11):4049–59. doi:10.1021/es050228s.
- Docherty, K. S., and P. J. Ziemann. 2003. Effects of Stabilized Criegee Intermediate and OH Radical Scavengers on Aerosol Formation from Reactions of β-Pinene with O3. Aerosol. Sci. Technol. 37 (11):877–91. doi:10.1080/02786820300930.
- Docherty, K. S., and P. J. Ziemann. 2006. Reaction of Oleic Acid Particles with NO3 Radicals: Products, Mechanism, and Implications for Radical-Initiated Organic Aerosol Oxidation. J. Phys. Chem. A. 110 (10):3567–77. doi:10.1021/jp0582383.
- Donahue, N. M., G. T. Drozd, S. A. Epstein, A. A. Presto, and J. H. Kroll. 2011. Adventures in Ozoneland: Down the Rabbit-Hole. Phys. Chem. Chem. Phys. 13 (23): 10848–57. doi:10.1039/c0cp02564j.
- Durham, L. J., C. F. Wurster, and H. S. Mosher. 1958. Peroxides. VIII. The Mechanism for the Thermal Decomposition of n-Butyl Hydroperoxide and n-Butyl 1-Hydroxybutyl Peroxide. J. Am. Chem. Soc. 80 (2):332–7. doi:10.1021/ja01535a020.
- Ehn, M., E. Kleist, H. Junninen, T. Petäjä, G. Lönn, S. Schobesberger, M. Dal Maso, A. Trimborn, M. Kulmala, D. R. Worsnop, et al. 2012. Gas Phase Formation of Extremely Oxidized Pinene Reaction Products in Chamber and Ambient Air. Atmos. Chem. Phys. 12 (11):5113–27. doi:10.5194/acp-12-5113-2012.
- Ehn, M., J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, et al. 2014. A Large Source of Low-Volatility Secondary Organic Aerosol. Nature. 506 (7489):476–9. doi:10.1038/nature13032.
- Epstein, S. A., S. L. Blair, and S. A. Nizkorodov. 2014. Direct Photolysis of α-Pinene Ozonolysis Secondary Organic Aerosol: Effect on Particle Mass and Peroxide Content. Environ. Sci. Technol. 48 (19):11251–8. doi:10.1021/es502350u.
- Friedel, R. A., and A. G. Sharkey. 1956. Mass Spectra of Acetal-Type Compounds. Anal. Chem. 28 (6):940–4. doi:10.1021/ac60114a004.
- Gao, S., M. Keywood, N. L. Ng, J. Surratt, V. Varutbangkul, R. Bahreini, R. C. Flagan, and J. H. Seinfeld. 2004. Low-Molecular-Weight and Oligomeric Components in Secondary Organic Aerosol from the Ozonolysis of Cycloalkenes and α-Pinene. J. Phys. Chem. A. 108 (46):10147–64. doi:10.1021/jp047466e.
- Gao, Y., W. A. Hall, and M. V. Johnston. 2010. Molecular Composition of Monoterpene Secondary Organic Aerosol at Low Mass Loading. Environ. Sci. Technol. 44 (20):7897–902. doi:10.1021/es101861k.
- Griesbaum, K., and J. Neumeister. 1982. Ozonolysis of Symmetrically 1,2-Disubstituted Ethylenes in HCl/Methanol Solutions: Acid Catalyzed Reactions of Primary Cleavage Products. Chem. Berichte-Recueil. 115:2697–706. doi:10.1002/cber.19821150803.
- Guo, H., J. Liu, K. D. Froyd, J. M. Roberts, P. R. Veres, P. L. Hayes, J. L. Jimenez, A. Nenes, and R. J. Weber. 2017. Fine Particle pH and Gas–Particle Phase Partitioning of Inorganic Species in Pasadena, California, During the 2010 CalNex Campaign. Atmos. Chem. Phys. 17 (9):5703–19. doi:10.5194/acp-17-5703-2017.
- Guo, H., L. Xu, A. Bougiatioti, K. M. Cerully, S. L. Capps, J. R. Hite Jr, A. G. Carlton, S.-H. Lee, M. H. Bergin, N. L. Ng, et al. 2015. Fine-Particle Water and pH in the Southeastern United States. Atmos. Chem. Phys. 15 (9):5211–28. doi:10.5194/acp-15-5211-2015.
- Hayes, M. J., and D. C. Pepper. 1961. The Solubility of H2SO4 in 1,2-Dichloroethane. Trans. Faraday Soc. 57:432–5. doi:10.1039/TF9615700432.
- Krapf, M., I. El Haddad, E. A. Bruns, U. Molteni, K. R. Daellenbach, A. S. H. Prévôt, U. Baltensperger, and J. Dommen. 2016. Labile Peroxides in Secondary Organic Aerosol. Chem. 1 (4):603–16. doi:10.1016/j.chempr.2016.09.007.
- Kristensen, K., Å. K. Watne, J. Hammes, A. Lutz, T. Petäjä, M. Hallquist, M. Bilde, and M. Glasius. 2016. High-Molecular Weight Dimer Esters are Major Products in Aerosols from α-Pinene Ozonolysis and the Boreal Forest. Environ. Sci. Technol. Lett. 3 (8):280–5. doi:10.1021/acs.estlett.6b00152.
- Lin, S. T., L. L. Tien, and N. C. Chang. 1989. Mass Spectra of Some Acetals. Analyst. 114 (9):1083–5. doi:10.1039/AN9891401083.
- Matsunaga, A., and P. J. Ziemann. 2009. Yields of β-Hydroxynitrates and Dihydroxynitrates in Aerosol Formed from OH Radical-Initiated Reactions of Linear Alkenes in the Presence of NOx. J. Phys. Chem. A. 113 (3):599–606. doi:10.1021/jp807764d.
- McGillen, M. R., B. F. E. Curchod, R. Chhantyal-Pun, J. M. Beames, N. Watson, M. A. H. Khan, L. McMahon, D. E. Shallcross, and A. J. Orr-Ewing. 2017. Criegee Intermediate-Alcohol Reactions, A Potential Source of Functionalized Hydroperoxides in the Atmosphere. Earth Space Chem. 1 (10):664–72. doi:10.1021/acsearthspacechem.7b00108.
- McLafferty, F. W. 2012. Mass Spectrometry of Organic Ions. 4th ed. Sausalito, CA: University Science Books.
- Mertes, P., L. Pfaffenberger, J. Dommen, M. Kalberer, and U. Baltensperger. 2012. Development of a Sensitive Long Path Absorption Photometer to Quantify Peroxides in Aerosol Particles (Peroxide-LOPAP). Atmos. Meas. Tech. 5 (10):2339–48. doi:10.5194/amt-5-2339-2012.
- Müller, L., M. C. Reinnig, J. Warnke, and T. Hoffmann. 2008. Unambiguous Identification of Esters as Oligomers in Secondary Organic Aerosol Formed from Cyclohexene and Cyclohexene/Α-Pinene Ozonolysis. Atmos. Chem. Phys. 8 (5):1423–33. doi:10.5194/acp-8-1423-2008.
- Mutzel, A., L. Poulain, T. Berndt, Y. Iinuma, M. Rodigast, O. Böge, S. Richters, G. Spindler, M. Sipila, T. Jokinen, et al. 2015. Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study. Environ. Sci. Technol. 49 (13):7754–61. doi:10.1021/acs.est.5b00885.
- Orlando, J. J., and G. S. Tyndall. 2012. Laboratory Studies of Organic Peroxy Radical Chemistry: An Overview with Emphasis on Recent Issues of Atmospheric Significance. Chem. Soc. Rev. 41 (19):6294–317. doi:10.1039/c2cs35166h.
- Peng, Z., D. A. Day, H. Stark, R. Li, J. Lee-Taylor, B. B. Palm, W. H. Brune, and J. L. Jimenez. 2015. HOx Radical Chemistry in Oxidation Flow Reactors with Low-Pressure Mercury Lamps Systematically Examined by Modeling. Atmos. Meas. Tech. 8 (11):4863–90. doi:10.5194/amt-8-4863-2015.
- Praske, E., R. V. Otkjaer, J. D. Crounse, J. C. Hethcox, B. M. Stoltz, H. G. Kjaergaard, and P. O. Wennberg. 2018. Atmospheric Autoxidation is Increasingly Important in Urban and Suburban North America. Proceed. Natl. Acad. Sci. USA. 115 (1):64–9. doi:10.1073/pnas.1715540115.
- Ranney, A. P., and P. J. Ziemann. 2016. Kinetics of Acid-Catalyzed Dehydration of Cyclic Hemiacetals in Organic Aerosol Particles in Equilibrium with Nitric Acid Vapor. J. Phys. Chem. A. 120 (16):2561–8. doi:10.1021/acs.jpca.6b01402.
- Ranney, A. P., and P. J. Ziemann. 2017. Identification and Quantification of Oxidized Organic Aerosol Compounds Using Derivatization, Liquid Chromatography, and Chemical Ionization Mass Spectrometry. Aerosol Sci. Technol. 51 (3): 342–53. doi:10.1080/02786826.2016.1271108.
- Riva, M., S. H. Budisulistiorini, Z. Zhang, A. Gold, J. A. Thornton, B. J. Turpin, and J. D. Surratt. 2017. Multiphase Reactivity of Gaseous Hydroperoxide Oligomers Produced from Isoprene Ozonolysis in the Presence of Acidified Aerosols. Atmos. Environ. 152:314–22. doi:10.1016/j.atmosenv.2016.12.040.
- Schwarzenbach, R. P., P. M. Gschwend, and D. M. Imboden. 2003. Environmental Organic Chemistry. Hoboken, NJ: JohnWiley & Sons, Inc.
- SDBSWeb. 2017. National Institute of Advanced Industrial Science and Technology. Accessed December. http://sdbs.db.aist.go.jp.
- Stein, S. E. 2016. Mass Spectra. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69, eds. P. J. Linstrom and W. G. Mallard. Gaithersburg, MD: National Institute of Standards and Technology.
- Tobias, H. J., P. M. Kooiman, K. S. Docherty, and P. J. Ziemann. 2000. Real-Time Chemical Analysis of Organic Aerosols Using a Thermal Desorption Particle Beam Mass Spectrometer. Aerosol Sci. Technol. 33 (1–2):170–90. doi:10.1080/027868200410912.
- Tobias, H. J., and P. J. Ziemann. 2000. Thermal Desorption Mass Spectrometric Analysis of Organic Aerosol Formed from Reactions of 1-Tetradecene and O3 in the Presence of Alcohols and Carboxylic Acids. Environ. Sci. Technol. 34 (11):2105–15. doi:10.1021/es9907156.
- Tobias, H. J., and P. J. Ziemann. 2001. Kinetics of the Gas-Phase Reactions of Alcohols, Aldehydes, Carboxylic Acids, and Water with the C13 Stabilized Criegee Intermediate Formed from Ozonolysis of 1-Tetradecene. J. Phys. Chem. A. 105 (25):6129–35. doi:10.1021/jp004631r.
- Vinnik, M. I., and P. A. Obraztsov. 1990. The Mechanism of the Dehydration of Alcohols and the Hydration of Alkenes in Acid Solution. Russ. Chem. Rev. 59 (1):63–77. doi:10.1070/RC1990v059n01ABEH003510.
- Wexler, A. S., and S. L. Clegg. 2002. Atmospheric Aerosol Models for Systems Including the Ions H+, NH4+, na+, SO42–, NO3–, cl–, br–, and H2O. J. Geophys. Res. 107 (D14):ACH14.1–ACH14.14. doi:10.1029/2001JD000451.
- Wurster, C. F., L. J. Durham, and H. S. Mosher. 1958. Peroxides. VII. The Thermal Decomposition of Primary Hydroperoxides. J. Am. Chem. Soc. 80 (2):327–331. doi:10.1021/ja01535a019.
- Zelikman, E. S., Y. N. Yur’ev, L. B. Berezova, and V. K. Tsyskovskii. 1971. Ozonolysis of Olefins in the Presence of Aliphatic Alcohols and Acids. Zhurnal Org. Khimii. 7:633–6.
- Zhang, Y., Y. Chen, A. T. Lambe, N. E. Olson, Z. Lei, R. L. Craig, Z. Zhang, A. Gold, T. B. Onasch, J. T. Jayne, et al. 2018. Effect of the Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX). Environ. Sci. Technol. Lett. 5 (3):167–74. doi:10.1021/acs.estlett.8b00044.
- Ziemann, P. J. 2002. Evidence for Low-Volatility Diacyl Peroxides as a Nucleating Agent and Major Component of Aerosol Formed from Reactions of O3 with Cyclohexene and Homologous Compounds. J. Phys. Chem. A. 106 (17):4390–402. doi:10.1021/jp012925m.
- Ziemann, P. J. 2003. Formation of Alkoxyhydroperoxy Aldehydes and Cyclic Peroxyhemiacetals from Reactions of Cyclic Alkenes with O3 in the Presence of Alcohols. J. Phys. Chem. A. 107 (12):2048–60. doi:10.1021/jp022114y.
- Ziemann, P. J., and R. Atkinson. 2012. Kinetics, Products, and Mechanisms of Secondary Organic Aerosol Formation. Chem. Soc. Rev. 41 (19):6582–605. doi:10.1039/c2cs35122f.